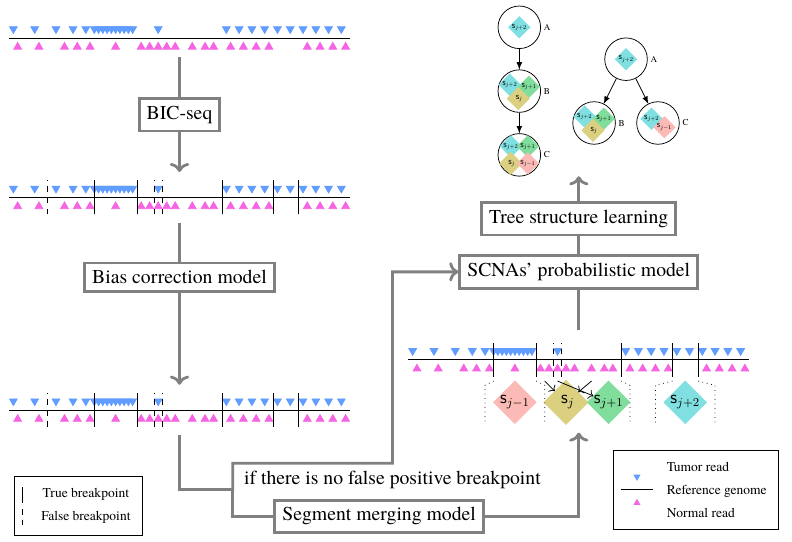
msphy-SCNAClonal is a SCNA's subclonal reconstruction pipeline based on multi-stage tree structure learning, which could infer the tree structure of tumor's evolution process from various kinds of sequencing data from a tumor's multiple samples, such as multiple samples' NGS data obtained at different time points, or, multiple NGS/Target sequencing/Single cell sequencing or their combination data.
seaborn
pymc
ete2
pydp
python2-backports.functools_lru_cache
gwpy
pysam
scipy
numpy
matplotlib
usage: run.py preprocess [-h] [--nBamName NBAMNAME]
[--tBamNameL TBAMNAMEL [TBAMNAMEL ...]]
[--bedNameL BEDNAMEL [BEDNAMEL ...]]
[--refFaName REFFANAME] [--pathPreFix PATHPREFIX]
[--subcloneNumL SUBCLONENUML [SUBCLONENUML ...]]
[--coverageL COVERAGEL [COVERAGEL ...]]
[--maxCopyNumber MAXCOPYNUMBER]
[--baselineThredLOH BASELINETHREDLOH]
[--baselineThredAPM BASELINETHREDAPM]
[--minDepth MINDEPTH] [--minBqual MINBQUAL]
[--minMqual MINMQUAL] [--processNum PROCESSNUM]
[--bedCorrectedPath BEDCORRECTEDPATH]
[--pklPath PKLPATH] [--answerFilePath ANSWERFILEPATH]
[--gcCorrectionMethod GCCORRECTIONMETHOD]
[--readFromBed READFROMBED] [--mergeSeg MERGESEG]
[--pklFlag PKLFLAG] [--isFixedC ISFIXEDC]
optional arguments:
-h, --help show this help message and exit
--nBamName NBAMNAME BAM file for normal sample.
--tBamNameL TBAMNAMEL [TBAMNAMEL ...]
BAM files for tumor samples sorted in chronological
order.
--bedNameL BEDNAMEL [BEDNAMEL ...]
BED files for segments of each sample in chronological
order.
--refFaName REFFANAME
FASTA file for reference genome.
--pathPreFix PATHPREFIX
Base name of the preprocessed input file to be
created.
--subcloneNumL SUBCLONENUML [SUBCLONENUML ...]
Set the subclone numbers
--coverageL COVERAGEL [COVERAGEL ...]
Set the coverage numbers
--maxCopyNumber MAXCOPYNUMBER
Set the maximum copy number
--baselineThredLOH BASELINETHREDLOH
baseline Thred of LOH
--baselineThredAPM BASELINETHREDAPM
baseline Thred of APM
--minDepth MINDEPTH Minimum reads depth required for both normal and tumor
samples. Default is 20.
--minBqual MINBQUAL Minimum base quality required. Default is 10.
--minMqual MINMQUAL Minimum mapping quality required. Default is 10.
--processNum PROCESSNUM
Number of processes to launch for preprocessing.
Default is 1.
--bedCorrectedPath BEDCORRECTEDPATH
The name of corrected BICseq result file
--pklPath PKLPATH Load the pkl path
--answerFilePath ANSWERFILEPATH
Load the answer file path
--gcCorrectionMethod GCCORRECTIONMETHOD
The gc correction method, one of auto and visual
--readFromBed READFROMBED
get read from Bed (True), from bam file if set it
False
--mergeSeg MERGESEG to merge segment or not to
--pklFlag PKLFLAG The pkl flag
--isFixedC ISFIXEDC Fix Copy number
usage: run.py model [-h] [-b WRITEBACKUPSEVERY] [-S WRITESTATEEVERY]
[-k TOPKTREES] [-f CLONALFREQS] [-B BURNINSAMPLENUM]
[-s MCMCSAMPLENUM] [-i MHITERATIONS] [-r RANDOMSEED]
[-t TMPDIR] [-p PARAMSFILE]
[--inputDataFile INPUTDATAFILE]
[--inputDataTextFile INPUTDATATEXTFILE]
[--isMerged ISMERGED] [--isCrossing ISCROSSING]
[--crossingFile CROSSINGFILE]
[--isSingleCell ISSINGLECELL]
[--singleCellFile SINGLECELLFILE]
[--maxCopyNumber MAXCOPYNUMBER] [--noTag NOTAG]
[--isParameterized ISPARAMETERIZED]
optional arguments:
-h, --help show this help message and exit
-b WRITEBACKUPSEVERY, --write-backups-every WRITEBACKUPSEVERY
Number of iterations to go between writing backups of
program state
-S WRITESTATEEVERY, --write-state-every WRITESTATEEVERY
Number of iterations between writing program state to
disk. Higher values reduce IO burden at the cost of
losing progress made if program is interrupted.
-k TOPKTREES, --top-k-trees TOPKTREES
Output file to save top-k trees in text format
-f CLONALFREQS, --clonal-freqs CLONALFREQS
Output file to save clonal frequencies
-B BURNINSAMPLENUM, --burnin-samples BURNINSAMPLENUM
Number of burnin samples
-s MCMCSAMPLENUM, --mcmc-samples MCMCSAMPLENUM
Number of MCMC samples
-i MHITERATIONS, --mh-iterations MHITERATIONS
Number of Metropolis-Hastings iterations
-r RANDOMSEED, --random-seed RANDOMSEED
Random seed for initializing MCMC sampler
-t TMPDIR, --tmp-dir TMPDIR
Path to directory for temporary files
-p PARAMSFILE, --params PARAMSFILE
JSON file listing run parameters, generated by the
parser
--inputDataFile INPUTDATAFILE
File listing data(SCNA data, either semgent or
stripe). For proper format, see README.md.
--inputDataTextFile INPUTDATATEXTFILE
Text file listing data(SCNA stripes). For proper
format, see README.md.
--isMerged ISMERGED is merged data file
--isCrossing ISCROSSING
using crossing file
--crossingFile CROSSINGFILE
The crossing file.
--isSingleCell ISSINGLECELL
using single cell file
--singleCellFile SINGLECELLFILE
The single cell file.
--maxCopyNumber MAXCOPYNUMBER
Max copy number
--noTag NOTAG to remove all tag or not
--isParameterized ISPARAMETERIZED
to make only one subpopulation in each time tag
usage: run.py postprocess [-h] [--treeFile TREEFILE]
[--SCNAPoolFile SCNAPOOLFILE]
[--answerFilePath ANSWERFILEPATH]
[--outputFolder OUTPUTFOLDER]
optional arguments:
-h, --help show this help message and exit
--treeFile TREEFILE File containing sampled trees
--SCNAPoolFile SCNAPOOLFILE
File containing SCNA pool
--answerFilePath ANSWERFILEPATH
Answer file path
--outputFolder OUTPUTFOLDER
Output folder