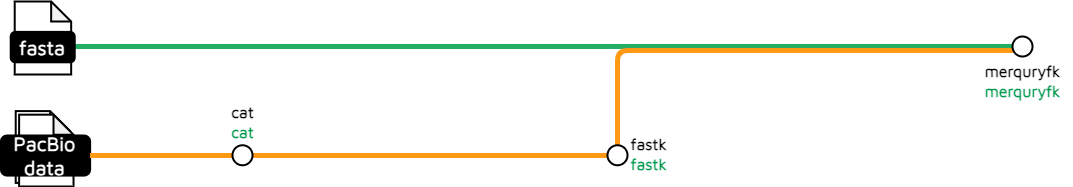
This uses merquryfk with data prepared using fastk.
workflow KMER {
take:
reference_tuple // Channel: [ val(meta), path(file) ]
reads_path // Channel: [ val(meta), val( str ) ]
main:
ch_versions = Channel.empty()
//
// LOGIC: PREPARE GET_READS_FROM_DIRECTORY INPUT
//
reads_path
.map { meta, reads_path ->
tuple(
[ id : meta.id,
single_end : true ],
reads_path
)
}
.set { get_reads_input }
//
// MODULE: GETS PACBIO READ PATHS FROM READS_PATH
//
ch_grabbed_read_paths = GrabFiles( get_reads_input )
//
// MODULE: JOIN PACBIO READ
//
CAT_CAT( ch_grabbed_read_paths )
ch_versions = ch_versions.mix( CAT_CAT.out.versions.first() )
//
// MODULE: COUNT KMERS
//
FASTK_FASTK( CAT_CAT.out.file_out )
ch_versions = ch_versions.mix( FASTK_FASTK.out.versions.first() )
//
// LOGIC: PREPARE MERQURYFK INPUT
//
FASTK_FASTK.out.hist
.combine( FASTK_FASTK.out.ktab )
.combine( reference_tuple )
.map{ meta_hist, hist, meta_ktab, ktab, meta_ref, primary ->
tuple( meta_hist, hist, ktab, primary, [] )
}
.set{ ch_merq }
//
// MODULE: USE KMER HISTOGRAM TO PRODUCE SPECTRA
//
MERQURYFK_MERQURYFK ( ch_merq )
ch_versions = ch_versions.mix( MERQURYFK_MERQURYFK.out.versions.first() )
CAT_CAT uses "conda-forge::pigz=2.3.4" and the call depends on the extensions:
// | input | output | command1 | command2 |
// |-----------|------------|----------|----------|
// | gzipped | gzipped | cat | |
// | ungzipped | ungzipped | cat | |
// | gzipped | ungzipped | zcat | |
// | ungzipped | gzipped | cat | pigz |
prefix = task.ext.prefix ?: "${meta.id}${file_list[0].substring(file_list[0].lastIndexOf('.'))}"
out_zip = prefix.endsWith('.gz')
in_zip = file_list[0].endsWith('.gz')
command1 = (in_zip && !out_zip) ? 'zcat' : 'cat'
command2 = (!in_zip && out_zip) ? "| pigz -c -p $task.cpus $args2" : ''
"""
$command1 \\
$args \\
${file_list.join(' ')} \\
$command2 \\
> ${prefix}
FASTTK_FASTTK uses a specific container ghcr.io/nbisweden/fastk_genescopefk_merquryfk:1.2 for merquryfk.
FastK is currently in PR state: galaxyproject/tools-iuc#5550 The module executes:
FastK \\
$args \\
-T$task.cpus \\
-M${task.memory.toGiga()} \\
-N${prefix}_fk \\
$reads
Merqury itself is already in the toolshed but Merqury.FK is missing and needs to be integrated. A conda package exist. MERQURYFK_MERQURYFK uses the same container to execute:
MerquryFK \\
$args \\
-T$task.cpus \\
${fastk_ktab.find{ it.toString().endsWith(".ktab") }} \\
$assembly \\
$haplotigs \\
$prefix