{kind=link}
{kind=link}
{kind=link}
A Galaxy user builds a workflow's tools, data flows and default parameter settings entirely through the GUI, compared to writing 100+ lines of custom DDL needed for this NF subworkflow described in the section that follows.
A preconfigured, shareable JBrowse viewer makes Galaxy workflow outputs immediately accessible, without the need to manually shuffle individual track files around.
A prototype repeat_density workflow is now available for testing and feedback.
Could this be usefully shown to NF users?
It could be created quickly because it did not require any new Galaxy tools, unlike many of the other remaining TreeVal subworkflows.

The server you are using must have the window_masker tool installed or this workflow will not work. Not yet an IUC tool so not on any usegalaxy servers yet.
In a Galaxy session, import this link to your Galaxy user workflows.
In a new history, upload a sample assembly GCA_031772095.1_chr1.fasta from this link from a nematode assembly as the input fasta to test the workflow
Execute the workflow with the nematode chromosome as input.
A new output JBrowse file with the repeats found as a track, will appear in the history.
View that JBrowse html history file. Note that the JBrowse tool must be whitelisted for HTML on the Galaxy server for this to work. Depending on the zoom level of JBrowse, you will see individual repeat sequences or for larger windows, JBrowse will estimate the density of repeats as a bar chart.
Zoomed in:

Zoomed out:

VGP and other interested researchers are asked to try it out on their own data to see if it is helpful. Please discuss, suggest improvements or raise issues here.
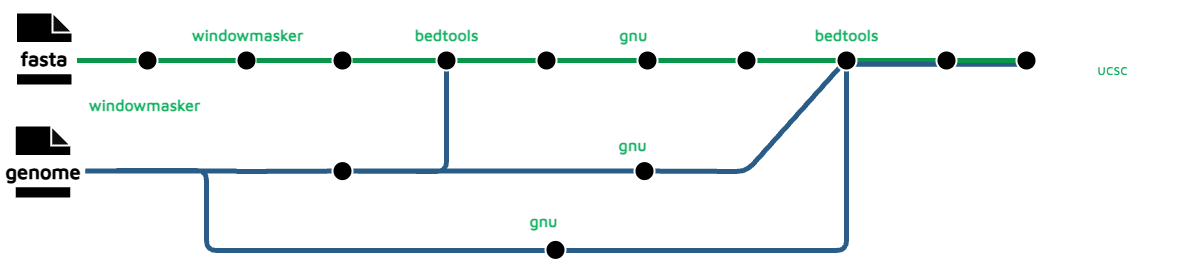
#!/usr/bin/env nextflow
//
// MODULE IMPORT BLOCK
//
include { WINDOWMASKER_USTAT } from '../../modules/nf-core/windowmasker/ustat/main'
include { WINDOWMASKER_MKCOUNTS } from '../../modules/nf-core/windowmasker/mk_counts/main'
include { EXTRACT_REPEAT } from '../../modules/local/extract_repeat'
include { BEDTOOLS_INTERSECT } from '../../modules/nf-core/bedtools/intersect/main'
include { BEDTOOLS_MAKEWINDOWS } from '../../modules/nf-core/bedtools/makewindows/main'
include { BEDTOOLS_MAP } from '../../modules/nf-core/bedtools/map/main'
include { RENAME_IDS } from '../../modules/local/rename_ids'
include { UCSC_BEDGRAPHTOBIGWIG } from '../../modules/nf-core/ucsc/bedgraphtobigwig/main'
include { GNU_SORT as GNU_SORT_A } from '../../modules/nf-core/gnu/sort/main'
include { GNU_SORT as GNU_SORT_B } from '../../modules/nf-core/gnu/sort/main'
include { GNU_SORT as GNU_SORT_C } from '../../modules/nf-core/gnu/sort/main'
include { REFORMAT_INTERSECT } from '../../modules/local/reformat_intersect'
include { REPLACE_DOTS } from '../../modules/local/replace_dots'
workflow REPEAT_DENSITY {
take:
reference_tuple // Channel: tuple [ val(meta), path(file) ]
dot_genome
main:
ch_versions = Channel.empty()
//
// MODULE: MARK UP THE REPEAT REGIONS OF THE REFERENCE GENOME
//
WINDOWMASKER_MKCOUNTS (
reference_tuple
)
ch_versions = ch_versions.mix( WINDOWMASKER_MKCOUNTS.out.versions )
//
// MODULE: CALCULATE THE STATISTICS OF THE MARKED UP REGIONS
//
WINDOWMASKER_USTAT(
WINDOWMASKER_MKCOUNTS.out.counts,
reference_tuple
)
ch_versions = ch_versions.mix( WINDOWMASKER_USTAT.out.versions )
//
// MODULE: USE USTAT OUTPUT TO EXTRACT REPEATS FROM FASTA
//
EXTRACT_REPEAT(
WINDOWMASKER_USTAT.out.intervals
)
ch_versions = ch_versions.mix( EXTRACT_REPEAT.out.versions )
//
// MODULE: CREATE WINDOWS FROM .GENOME FILE
//
BEDTOOLS_MAKEWINDOWS(
dot_genome
)
ch_versions = ch_versions.mix( BEDTOOLS_MAKEWINDOWS.out.versions )
//
// LOGIC: COMBINE TWO CHANNELS AND OUTPUT tuple(meta, windows_file, repeat_file)
//
BEDTOOLS_MAKEWINDOWS.out.bed
.combine( EXTRACT_REPEAT.out.bed )
.map{ meta, windows_file, repeat_meta, repeat_file ->
tuple (
meta,
windows_file,
repeat_file
)
}
.set { intervals }
//
// MODULE: GENERATES THE REPEAT FILE FROM THE WINDOW FILE AND GENOME FILE
//
BEDTOOLS_INTERSECT(
intervals,
dot_genome
)
ch_versions = ch_versions.mix( BEDTOOLS_INTERSECT.out.versions )
//
// MODULE: FIXES IDS FOR REPEATS
//
RENAME_IDS(
BEDTOOLS_INTERSECT.out.intersect
)
ch_versions = ch_versions.mix( RENAME_IDS.out.versions )
//
// MODULE: SORTS THE ABOVE BED FILES
//
GNU_SORT_A (
RENAME_IDS.out.bed // Intersect file
)
ch_versions = ch_versions.mix( GNU_SORT_A.out.versions )
GNU_SORT_B (
dot_genome // Genome file - Will not run unless genome file is sorted to
)
ch_versions = ch_versions.mix( GNU_SORT_B.out.versions )
GNU_SORT_C (
BEDTOOLS_MAKEWINDOWS.out.bed // Windows file
)
ch_versions = ch_versions.mix( GNU_SORT_C.out.versions )
//
// MODULE: ADDS 4TH COLUMN TO BED FILE USED IN THE REPEAT DENSITY GRAPH
//
REFORMAT_INTERSECT (
GNU_SORT_A.out.sorted
)
ch_versions = ch_versions.mix( GNU_SORT_C.out.versions )
//
// LOGIC: COMBINES THE REFORMATTED INTERSECT FILE AND WINDOWS FILE CHANNELS AND SORTS INTO
// tuple(intersect_meta, windows file, intersect file)
//
REFORMAT_INTERSECT.out.bed
.combine( GNU_SORT_C.out.sorted )
.map{ intersect_meta, bed, sorted_meta, windows_file ->
tuple (
intersect_meta,
windows_file,
bed
)
}
.set { for_mapping }
//
// MODULE: MAPS THE REPEATS AGAINST THE REFERENCE GENOME
//
BEDTOOLS_MAP(
for_mapping,
GNU_SORT_B.out.sorted
)
ch_versions = ch_versions.mix( BEDTOOLS_MAP.out.versions )
//
// MODULE: REPLACES . WITH 0 IN MAPPED FILE
//
REPLACE_DOTS (
BEDTOOLS_MAP.out.map
)
ch_versions = ch_versions.mix( REPLACE_DOTS.out.versions )
//
// MODULE: CONVERTS GENOME FILE AND BED INTO A BIGWIG FILE
//
UCSC_BEDGRAPHTOBIGWIG(
REPLACE_DOTS.out.bed,
GNU_SORT_B.out.sorted.map { it[1] } // Pulls file from tuple of meta and file
)
ch_versions = ch_versions.mix( UCSC_BEDGRAPHTOBIGWIG.out.versions )
emit:
repeat_density = UCSC_BEDGRAPHTOBIGWIG.out.bigwig
versions = ch_versions.ifEmpty(null)
}
First step, WINDOWMASKER_MKCOUNTS uses an NF core module to run a tool already available in the toolshed with this command line:
windowmasker -mk_counts \\
$args \\
-mem ${memory} \\
-in ${ref} \\
-out ${prefix}.txt
That repository also includes the command used in the next ustat step.
windowmasker -ustat \\
${counts} \\
$args \\
-in ${ref} \\
-out ${output}
https://github.com/goeckslab/WindowMasker may be useful here.
extract_repeats runs a perl script in the /tree/bin directory
extract_repeat.pl $file > ${prefix}_repeats.bed
so a new simple tool is needed to do:
#!/usr/local/bin/perl
use strict;
my $file = shift;
open(IN,$file);
my $last;
while (<IN>) {
if (/\>(\S+)/) {
$last = $1;
}
elsif (/(\d+)\s+-\s+(\d+)/) {
print "$last\t$1\t$2\n";
}
else {
die("Eh? $_");
}
}
Then there are calls to makewindows
bedtools \\
makewindows \\
${arg_input} \\
${args} \\
> ${prefix}.bed
and intersect
bedtools \\
intersect \\
-a $intervals1 \\
-b $intervals2 \\
$args \\
$sizes \\
> ${prefix}.${extension}
that can probably be done with existing toolshed bedtools.
rename_ids is a simple sed operation to replace a period with 0 - replace_dots below does the same thing.
$/
cat "${file}" \
| sed 's/\./0/g' > ${prefix}_renamed.bed
so could use the sed tool or a new specialised tool.
reformat_intersect is a local NF module that does
$/
cat "${file}" \
| awk '{print $0"\t"sqrt(($3-$2)*($3-$2))}'\
| sed 's/\./0/g' > ${prefix}.bed
so a new tool perhaps. Odd that it does replace_dots sed - is it redundant below?
Bedtools map does:
bedtools \\
map \\
-a $intervals1 \\
-b $intervals2 \\
$args \\
$sizes \\
> ${prefix}.${extension}
so can use the existing bedtools tools.
replace_dots (might be redundant give reform_intersect above but) calls the following sed command:
$/
cat "${file}" | sed 's/\./0/g' > "${prefix}_nodot.bed"
so a new tool will be needed or the IUC sed tool perhaps.
ucsc_bedgraphtobigwig calls
bedGraphToBigWig \\
$bedgraph \\
$sizes \\
${prefix}.bigWig
There is an existing bedgraph2BigWig wrapper: https://usegalaxy.eu/root?tool_id=wig_to_bigWig