| date | comments |
|---|---|
2023-04-07 |
true |
这里记录每周值得分享的生信相关内容,周日发布。
本杂志开源(GitHub: openbiox/weekly),欢迎提交 issue,投稿或推荐生信相关内容。
2022 年随着又一年的疫情走过去了,我们一起迎来了 2023 年的第一天。从去年末开始,我们国家经历新的变化,我们的生活也在经历新的转机。在这个阵痛时刻,我们辞旧迎新、复盘过去、期待将来。我想说,好的未来,就是坚持的现在。在过去的一年里,我们成立专门的周刊运维团队,坚持贡献着归纳和分享,向大家传递知识和更多的可能性。感谢各位读者的关注,也衷心感谢一些读者的喜欢,希望这里的一点微光,能在某个时间照亮你未曾可及的角落,那就是这里坚守的意义。
向所有奉献在一线防疫工作的人员致敬!🫡
向所有过去一年在努力活着的家庭致敬!🫡
向在人生中寻寻觅觅的你致敬!🫡
—— 王诗翔

首个“虫洞”登上 Nature 封面(图来源)。
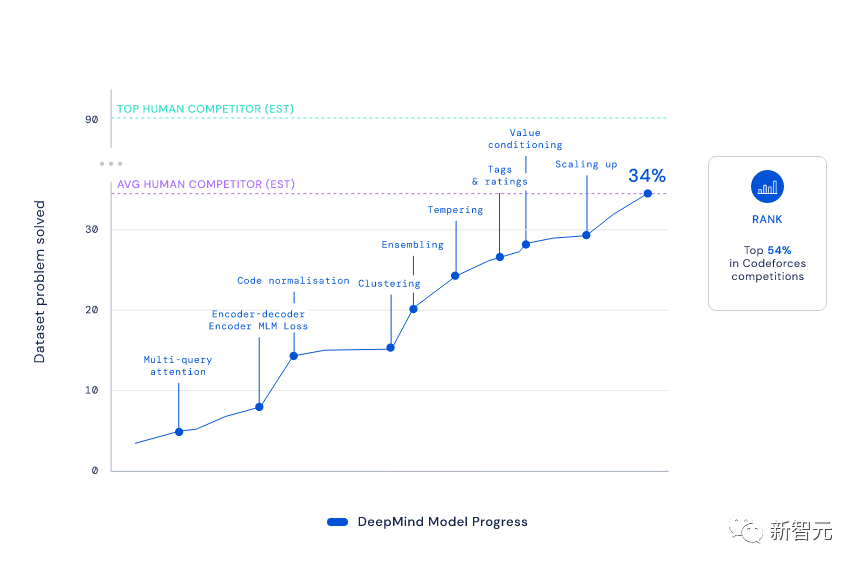
程序员中非常流行这样一种测试——编程竞赛。在竞赛中,主要考察的就是程序员通过经验进行批判性思维,为不可预见的问题创建解决方案的能力。这体现了人类智能的关键,而机器学习模型,往往很难模仿这种人类智能。但DeepMind的科学家们,打破了这一规律。
YujiA Li等人,使用自监督学习和编码器-解码器转换器架构,开发出了AlphaCode。它就在著名的Codeforces上,悄悄地参加了10场编程比赛,并一举击败了半数的人类码农。
@ShixiangWang - AI 正在为生活和科技工作创造更多的可能性。作为研究者,我们需要尽力去利用技术武装自己,提高效率;同时也要关注技术给我们带来的复杂性、诱导性,避免偏离我们实际所追寻的目标。
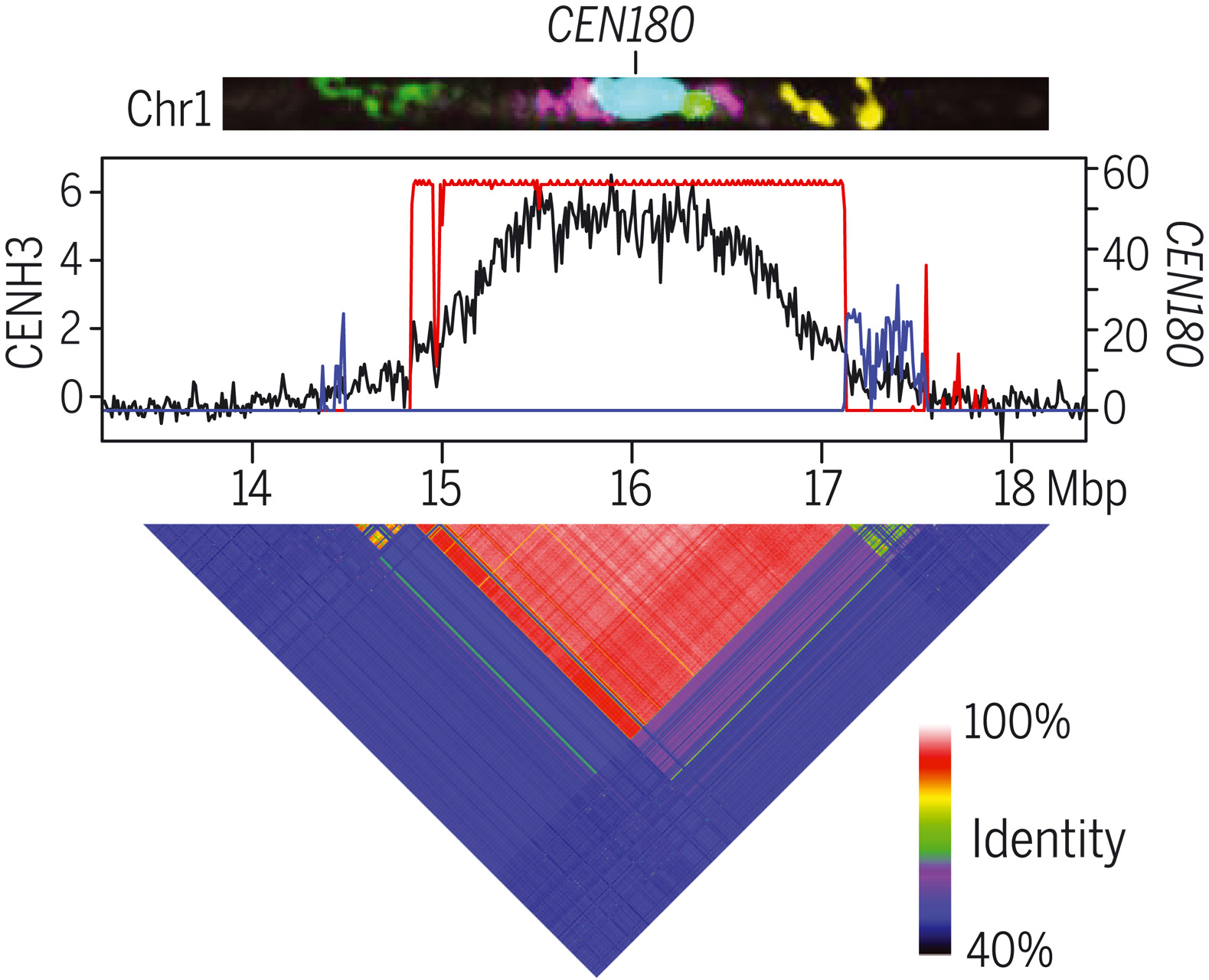
T2T测序在动物领域比植物领域应用更广泛。本文成功组装了拟南芥5条染色体着丝粒的首个全序列基因组Col-CEN,揭示了拟南芥着丝粒的遗传和表观遗传图景。
2、Nature Communication | 基于全基因组测序数据定制癌症疗法,基因组特征模型iGenSig助力精准医疗临床决策支持

美国UPMC希尔曼癌症中心的研究团队开发了一组用于预测癌症药物反应的基因组特征模型iGenSig,并使用独立的细胞系和临床数据集对该模型进行了验证。该团队引入了一种白盒方法,称为整合基因组特征分析(iGenSig)(图1),利用高维冗余基因组特征作为整合基因组特征,以增强基于多组学的精准肿瘤学建模的可转移性。iGenSig方法旨在解决基于大数据建模的透明性、跨数据集适用性和可解释性问题。在临床试验数据集的交叉适用性方面,iGenSig模型表现出了更好的性能,能够容忍基因组数据中的实验变异和偏差。iGenSig模型可以在每一个详细的步骤中进行管理,利用研究团队开发的富集分析方法,可以很容易地从生物学角度解释基因组特征的潜在通路。研究团队期望,作为基于大数据的建模方法,iGenSig将在药物基因组学和临床试验数据集的治疗反应建模中有广泛应用。
3、iMeta | iMetaLab Suite-宏蛋白组学分箱一站式工具箱

宏蛋白组学研究所需的生物信息学工作流程包含了蛋白质鉴定(搜库)、定量至下游功能和分类分析,一定程度上限制了其他领域微生物研究人员对宏蛋白组学的使用和探索,为此,本工作开发了涵盖宏蛋白组中最常用的功能、分类和统计分析的流程化工具箱。
4、Nature Communication | 如何测量复杂网络的维度?
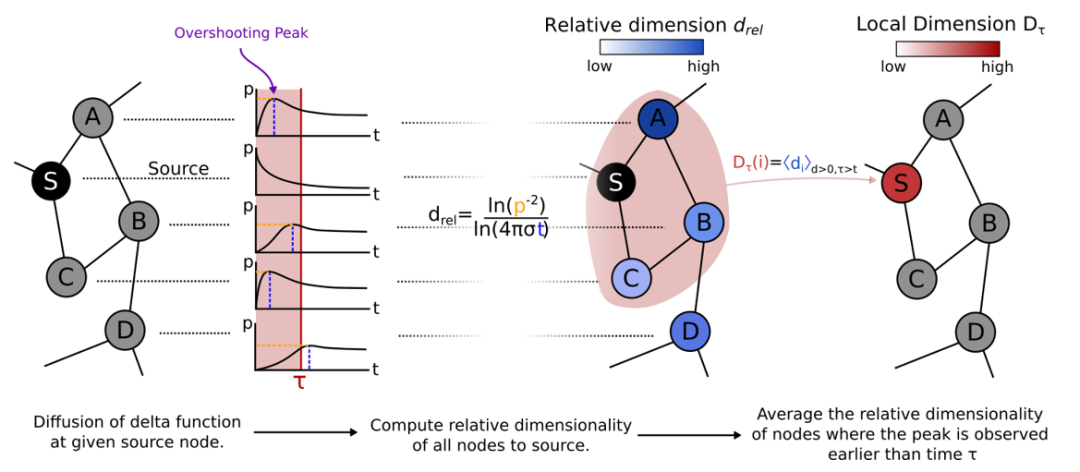
本研究提出通过扩散动力学过程来探测空间几何并定义维度的新方法,空间中的每个点都可以被分配一个相对于扩散源的相对维度(relative dimension)。这一概念为网络的局部和全局维度提供了依赖于尺度的定义,能反映网络的拓扑属性。为了展示相对维度概念可以在现实复杂系统应用,作者还展示了维度测量在网络传播动态过程的应用。
在2022年伦敦会议上,Nanopore technologies宣布Guppy已更新到v6.1.1。Remora是一款位于Minion的新软件,正式推荐用于检测DNA修饰。
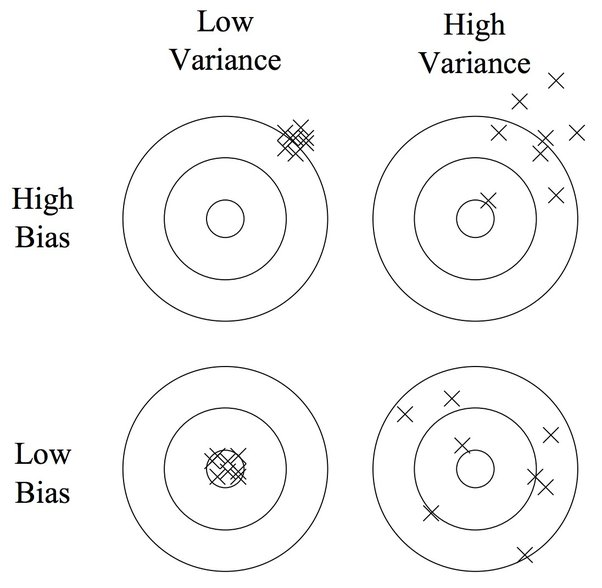
bias-variance tradeoff 是机器学习的核心概念,本帖子展示了大家对于这一概念的理解和讨论。
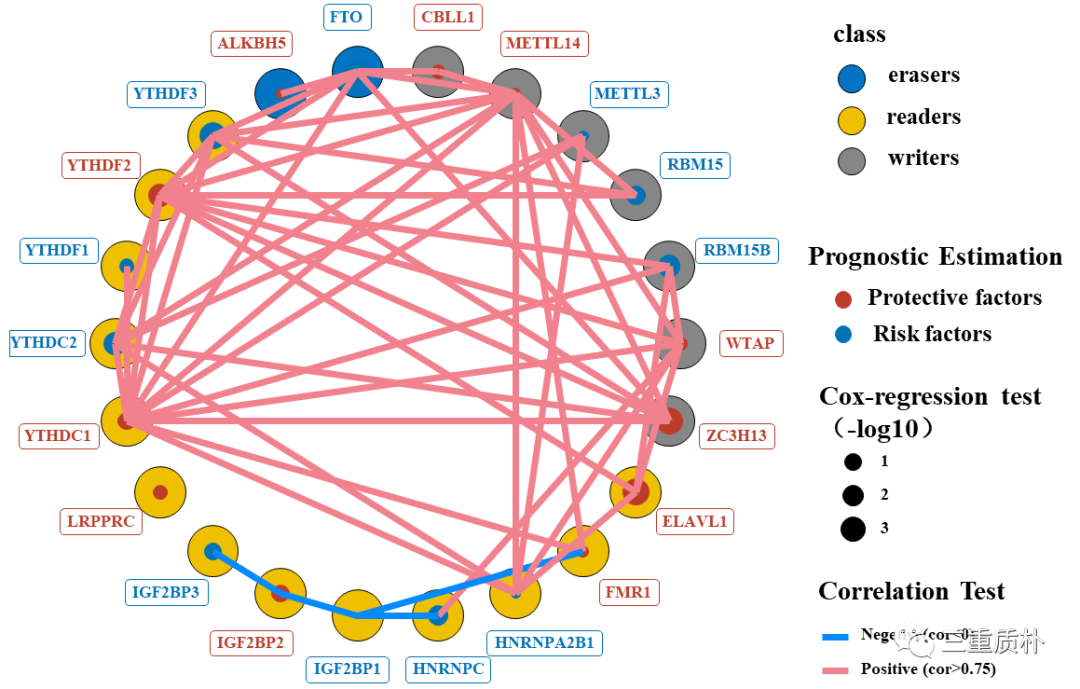
本文以TCGA-THCA数据为例,介绍了如何将基因相关性、cox风险和归类属性在同一张图中进行展示。

该文介绍了如何绘制textbox(文本框)并且如何将其作为热图的annotation。
hello = {
exec "echo Hello"
}
world = {
exec "echo World"
}
Bpipe.run { hello + world }许多从事数据科学工作的人最终以shell脚本的形式运行作业。虽然这使得运行它们很容易,但也有很多限制。例如,当脚本中途失败时,通常很难判断失败的位置或原因,从失败点重新启动作业就更难了。没有执行命令或控制台输出的自动日志,以确保以后可以看到发生了什么。有时作业会在中途失败,留下创建一半的文件,可能会与好文件混淆。修改管道也很耗时,而且容易出错——添加或删除一个步骤需要在多个地方进行修改,在一个地方更改文件名很容易导致后面的命令失败,甚至更糟,在不正确的数据上运行。Bpipe试图解决所有这些问题(甚至更多!),同时尽可能少地偏离shell脚本的简单性。事实上,你的Bpipe脚本通常看起来与您开始时使用的原始shell脚本非常相似。


GREAT包是一个直接对基因区域进行功能富集分析的R包,分为本地分析和在线分析。

核心功能:broken()函数,输入一组观测指标和一个线性模型或逻辑回归模型,输出这些指标对模型预测结果的贡献。

stLearn支持空间转录组数据的细胞聚类、细胞轨迹分析、细胞-细胞相互作用以及可视化等功能。
- 教程:https://stlearn.readthedocs.io/en/latest/index.html
- github:https://github.com/BiomedicalMachineLearning/stLearn
13、bigstatsr | 用于存储在磁盘上的大矩阵的统计工具R包

很多情况下基因组学数据由于所需内存太大而无法载入 R 进行处理。本包提供对编码为矩阵的大规模数据进行快速统计分析的功能。由于内存映射到磁盘上的二进制文件,这个包可以处理太大而无法装入内存的矩阵。
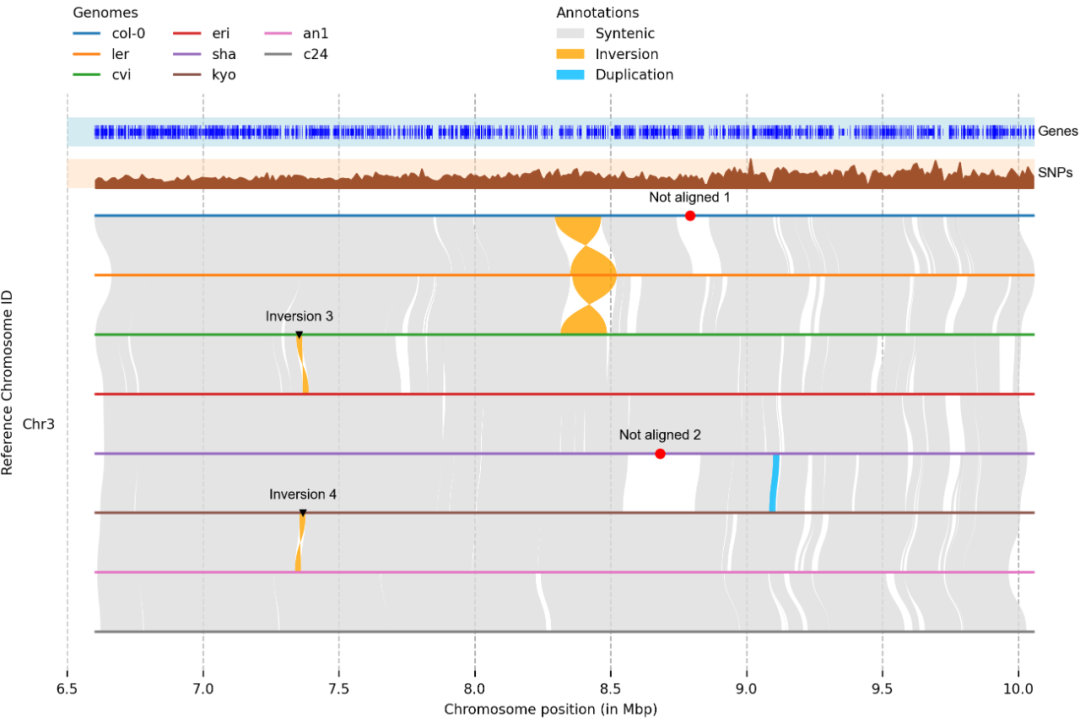
一个有效的可视化基因组之间结构变异和重排的有效工具,并可以用于在染色体水平上比较基因组或放大任何选定区域。
- GitHub: github.com/schneebergerlab/plotsr
16、视频 | David Liu 教授介绍无需打断 DNA 双链的基因编辑
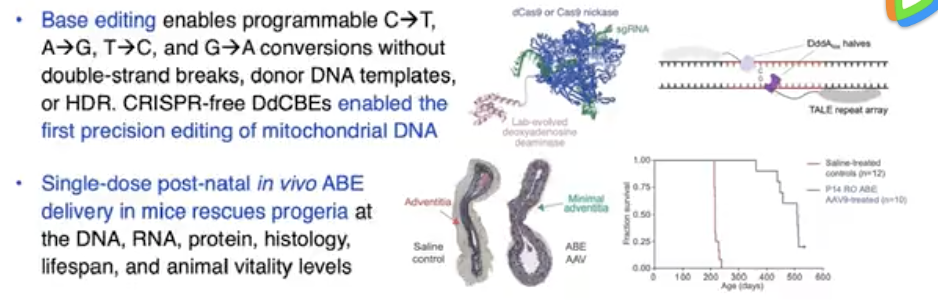
David Liu (美籍华裔,中文名:刘如谦)是哈佛大学的教授,专攻方向是基因编辑。
David Liu 在这个视频中介绍了新的碱基编辑方法、和 prime 编辑方法。
这两种基因编辑的方法,都避免了用 CRISPR 方法会导致的 DNA 双链断裂。
- 2022年2月:第19期:2022年值得关注的7大前沿技术
「Openbiox 生信周刊」运维小队:
@ShixiangWang(王诗翔)@kkjtmac(阚科佳)@NiEntropy(赵启祥)@He-Kai-fly(何凯)@JnanZhang(张佳楠)@Tomcxf(陈啸枫)@wangdepin(王德品)
这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。
微信搜索“优雅R”或者扫描二维码,即可订阅。

(完)