大学共同利用機関法人 情報・システム研究機構
データサイエンス共同利用基盤施設
ライフサイエンス統合データベースセンター
仲里 猛留
[email protected]
twitter: @chalkless
2019年11月29日(金) 東京農業大学 世田谷キャンパス
AJACS世田谷 > 次世代シーケンサー(NGS)と関連するデータベースツール
- 言葉
- 次世代シーケンサ
- 次世代シーケンサー
- 新型シーケンサ
- New-generation Sequencing (NGS)
- Next-generation Sequencing (NGS)
- 他にmassively parallel DNA sequencing とか...
- 最近は、 High-throughput DNA sequencing (technology) をよく使う印象(略語はNGS)
- 90年代
- ゲル板
- ポリアクリルアミドゲル電気泳動 + 蛍光標識ダイデオキシヌクレオチド

-
00年代
- キャピラリ
 ABI PRISM® 3100-Avant Genetic Analyzerより
ABI PRISM® 3100-Avant Genetic Analyzerより
- キャピラリ
-
10年代
- NGSの登場
- Sanger法(dideoxy法)→ パイロシーケンシング
- (参考)[原理の動画 (Illumina)](http://www.youtube.com/watch?v=l99aKKHcxC4)
次世代シーケンス解析サービス:概要・原理 | 北海道システム・サイエンス株式会社より
- ようするに顕微鏡+インターバル撮影/タイムラプス撮影 -インターバル撮影/タイムラプス撮影: http://www.youtube.com/watch?v=1Az1YX3GgDw
- 超並列
- どんなの?
-
Illumina HiSeq
-
Illumina MiSeq
-
PacBio
-
Ion Torrent
-
Togo picture gallery ( http://togotv.dbcls.jp/ja/pics.html ) より
-

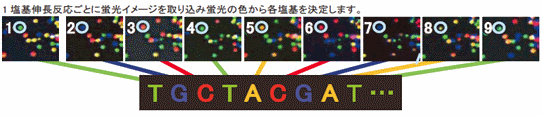




© 2011 DBCLS Licensed under CC 表示 2.1 日本 ←クレジットをいれれば、転載・改変・再利用 OK
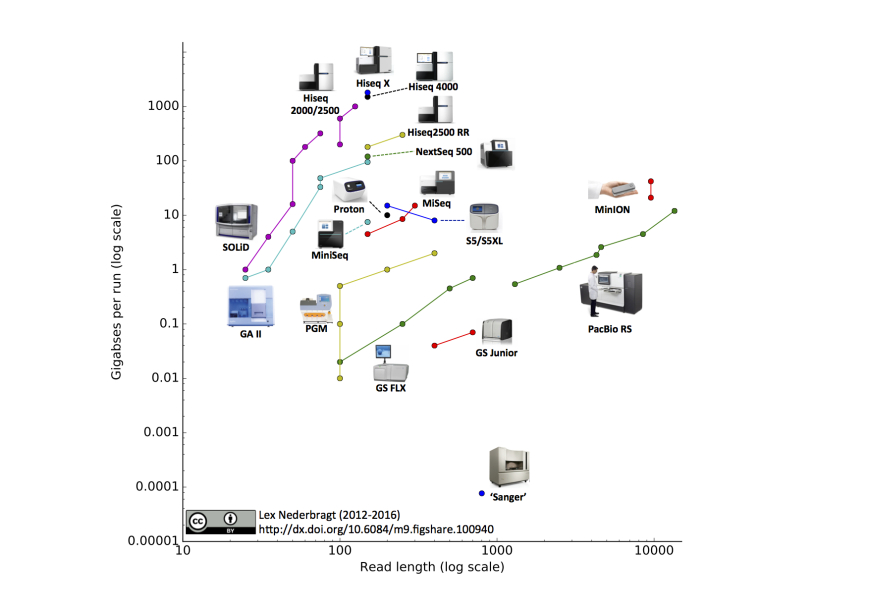 2016年7月時点でのシーケンサー各社の公称スペックをプロットしたもの (引用: Developments in high throughput sequencing – July 2016 edition, https://flxlexblog.wordpress.com/2016/07/08/developments-in-high-throughput-sequencing-july-2016-edition/ )。
2016年7月時点でのシーケンサー各社の公称スペックをプロットしたもの (引用: Developments in high throughput sequencing – July 2016 edition, https://flxlexblog.wordpress.com/2016/07/08/developments-in-high-throughput-sequencing-july-2016-edition/ )。
- 【実習】どのくらいのデータ量になるか考えてみましょう
- ゲル板:750 (base/lane) × 48/4 lanes = 9kbase
- キャピラリ:500 (base/lane) × 96 lane = 48kbase
- 次世代: 36 (base/seq) × 300M seq/run = 10.8Gbase = 10,800,000kbase
- ↑これらの数字は規模感をつかむだけなので、ざっくりな数字になっています(1 runにかかる時間は比較してないですし)
- ↑これらの数字は「塩基数」であって、シーケンサの出力である「画像データ」のデータサイズでないことに注意!
- そして、その画像データはSRAには登録されていない
- ゲノム、発現解析、メタゲノム解析、ChIP-Seq(転写因子解析)、SNP解析、...
- 目的によって必要なデータ量も違う

- 機器に合った利用を

- 目的によって必要なデータ量も違う
- Sequence Read Archive (SRA) というデータベース。NCBI(アメリカ)、EBI(ヨーロッパ)、DDBJ(日本)の3つでデータ交換をしている。
- SRA (NCBI)… 例
- ENA (European Nucleotide Archive, EBI)… 例
- DRA (DDBJ sequence read archive)… 例
- 試しに検索してみましょう: http://trace.ddbj.nig.ac.jp/DRASearch/ (キーワードで検索 → TypeをStudyで絞り込み。生物種でも)
- DBCLS SRA (DBCLS) … いろいろな観点(目的、機器、生物種)からの絞り込みが可能。改修+DDBJと統合中
- 文献からの検索
- 疾患からの検索・頻度・階層構造表示
- 生物種からの絞り込み例 … 種→亜種・株、種→属→科の検索も可能

-
参考図書・その1 〜 実験もやる人向け

-
参考図書・その2 〜 解析を詳しく

-
詳細な解析をひととおり知りたい
 https://biosciencedbc.jp/human/human-resources/workshop/h28-2
https://biosciencedbc.jp/human/human-resources/workshop/h28-2 -
解析について詳細な情報を探したい
 http://www.iu.a.u-tokyo.ac.jp/~kadota/r_seq.html
http://www.iu.a.u-tokyo.ac.jp/~kadota/r_seq.html -
解析環境・コマンドラインベース
 http://www.iu.a.u-tokyo.ac.jp/~kadota/r_seq.html#bioinfo_ngs_sokushu_2016_20160719
http://www.iu.a.u-tokyo.ac.jp/~kadota/r_seq.html#bioinfo_ngs_sokushu_2016_20160719
-
クオリティチェックには主にFASTQCというツールが使われます。
トリミングはさまざまなツールがあります(今回はtrim_galoreで例を示しています) -
入力データ:FASTQフォーマット

-
[参考] FASTAフォーマット

-
場合によっては(NCBIからダウンロードしたときなど)サイズ削減などのため、sra形式で圧縮されている場合があります。そのときはsra-toolkitでFASTQファイルを取り出したりします
-
コマンド例
- クオリティチェック
$ fastqc --nogroup -o DRR1234567.fastq- トリミング
trim_galore --paired --illumina --fastqc -o trimmed/ DRR1234567.R1.fastq DRR1234567.R2.fastq -
結果例

- HTMLファイルができると思ってください
- 今回、発現解析のところで用いるデータ(デスクトップ>AJACS_OWARI)の中にも実際のものがあります
- DBCLS SRAではあらかじめFastQCをかけた結果を表示できるように随時、処理をしています(自分でやらなくてよい!)
- 例: http://sra.dbcls.jp/search/view/SRR067385
-
ゲノムなどのリファレンス配列にNGSデータをマッピングします
-
bowtie、tophat2などなどさまざまなツールがあります
-
発現解析程度なら速度重視、SNP解析なら精度重視とツールも変わります
-
名前が違うだけで、中身は複数のツールの組み合わせということも多々あります
-
コマンド例:マッピング
hisat2 -f -p 24 -x genome.idx -U DRR1234567.trimmed.fasta -S results.sam -
結果:sam/bamフォーマット

-
コマンド例:形式変換
-
マッピングはsam形式かbam形式で出力されます
samは人間が読めるがサイズが大きいです。bamはプログラムで扱えるように(サイズを小さくするためにも)なっていますが人間には読めませんsamtools view -Sb SRR1294107.sam -o SRR1294107.bam(SAMからBAMへの変換)samtools view -h SRR1294107.bam -o SRR1294107.sam(BAMからSAMへの変換)
-
特に非モデル生物を扱う場合など、ゲノム情報がない場合は、RNA-Seqの発現データをリードだけでつなぎtranscriptを得る
-
Trinity を用いる
-
コマンド例
$ Trinity --seqType fq --left DRR1234567.R1.fastq --right DRR1234567.R2.fastq --max_memory 24G --CPU 16 -
結果例
$ head trinity_out_dir/Trinity.fasta >TRINITY_DN0_c0_g1_i1 len=390 path=[735:0-389] [-1, 735, -2] AAACTTCATAGATGAAATAAATGCTCATATACTATGTAGAAAATCTCCACATATATAAAA CAAAACATTTTGCTTTAAAACAGATATGATCACTAGGTGCAATGGCCTAATTCCCTGGCT … -
DNxxx_cXXX がクラスタとしてまとまったもの。-gXXXの部分が遺伝子、_iXXXがisoformに対応
- 発現量解析
- 遺伝子機能アノテーション
- BLASTで類似性のある遺伝子を検索
- hmmerでドメインサーチ
- → Trinityの関連でのアノテーションづけのページが非常に役立つ
https://trinotate.github.io/
-
基本的にはmappingと同様。ただし、1塩基のずれも重要なので(場合によってはmappingでのツールで大雑把にアラインメントをとった後)精度重視でのmappingを行う。その後、variantの解析を行う。
-
結果:vcfデータ(variant call format)

- ChIP-Atlas: http://chip-atlas.org/ (九州大・沖さん) 使い方動画もここにあり
- MircobeDB.jp: http://microbedb.jp/
AJACS世田谷 > 次世代シーケンサー(NGS)と関連するデータベースツール