Developed by Joseph Mahon and Adriana Toutoudaki during the Programming Week in November 2018 at the University of Manchester
The user requirements provided during the practical sessions were to create an application that could:
- Parse LRG XML files
- Extract Exon locations
As the tool is being designed to be used in a clinical environment, extra functionality was included to improve ease of use, safety and tracability.
- Automatic download of LRG XML file when provided with a HGNC ID.
- Extraction of genome build and transcripts from LRG XML to ensure accurate mapping
- Fully labelled BED files and BED data rows for traceability and audit purposes.
- Use of flags to allow integration within a pipeline or automated workflow.
- User interface to allow simple use by staff with limited command line experience.
Ensuring that software is developed according to best practice guidelines and testing is performed is essential when working in a clinical environment. Code that has not been written well, checked, reviewed and documented can pose a hazard to patient safety. Documentation availability, traceability, validation and verification are all important. These are also investigated by Laboratory accreditation body UKAS when assessing conformity with ISO15189 and so compliance is essential to ensure successful accreditation. The ACGS Guidelines for Development and Validation of Software have also been followed. The application has been developed using the PEP8 style guidelines to ensure that code is clearly formatted and readable.
Version control software (git) has been used to enable collaborative working in a safe and structured manner. It allows code review to be performed easily, and for an issues to be flagged or rolled back. Commits have been made in a granular manner, so that individual unwanted changes can be rolled back without affecting changes that are still required.
Some development decisions were made after seeking feedback from potential users. For example, the option to include introns or not was created after speaking with a Clinical Scientist about whether that would be a useful feature. This shows how feedback is important to ensure that the tools created are useful to the end user.
The application is compatible with Python 3 only. This decision was made as Python 2 will be unsupported in the near future.
It has been tested on both macOS and ubuntu-based linux operating systems. The program does run on Windows 7 but complete compatibility has not been tested.
Documentation is important when creating any software application, but is essential for software in a clinical environment. Knowing how to use a tool, as well as its limitations, output formats and other details are necessary for safe use. The output format of this tool and instructions on it's use are documented in this README file. Other documentation for code reviewers and developers is maintained within the codebase itself. Docstrings have been used to provide information on the roles of functions, and line comments provide supplementary information about short sections of code.
It is essential when writing software in a clinical environment that any data used or created is recorded to provide a trackable audit trail. To ensure that this trail is made, all BED files created by this tool are saved with a filename that includes all appropriate information:
HGNC Gene ID, LRG ID, Genome Version, Transcript, whether introns are included, the size of any flanking region and the date and time of file generation.
This information is also included within the header of the LRG file to ensure that it is visible when loaded into a genome browser. Each row in the resulting BED file makes use of the extended BED format and includes the additional field ‘name’, which is used as a descriptor for the row to allow identification. Retaining this information in the filename and BED header is important for audit purposes.
Awareness of indexing and formats is also essential to creating a tool which provides a safe to use output.
BED format uses 0-based numbering, whereas the LRG specification uses 1- based numbering (LRG Specification). BED format also does not include the chromEnd base when displaying the feature. This means that for an example LRG region of 3-7, the BED equivalent will be 2-7. The logic for this is that each index moves backwards by 1 place to accomodate the 0-based numbering to give 2-6, but 1 is added back onto the end of the range to allow the last base to be displayed. This is shown in the image below.

The tool has been written so that it can be run with a set of flags, which provide all of the information required to create a BED file. The minimum flags necessary for automated BED generation is one defining which LRG to use (by providing either an LRG ID, HGNC Gene Name, or LRG XML file), along with a transcript and genome assembly version. If one of these flags is missing, the user interface (UI) will display and prompt the user for their choice. Having this information is essential, as creating a BED file without defining a transcript is unsafe, especially in a clinical environment.
There are two other optional flags. These are not required for automated BED generation, and allow the user to specify whether they want to include intronic regions or add a flank to each region.
The program can also be run with no flags at all, which uses a terminal based UI to collect the information. If a user also provides flags which are incomplete, the UI will prompt the user for the missing items. The tool will not output a BED file unless the minimum safe information is provided. Flag use is detailed in Program Use below.
When planning the development, multiple python packages were investigated to fulfil particular functions. For example, lxml is a package which builds upon the functionality offered by the etree.ElementTree package. It provides some enhanced functionality and a slightly simpler interface, but as it is not included in the python standard library it requires installation. This is generally straightforward, but compatibility issues can arise when using different operating systems, such as Windows, where lxml cannot be installed solely using pip.
As the xml processing performed by this tool is simple, it was determined that using etree.ElementTree was sufficient and the compatibility and external package management overhead required for lxml was unnecessary.
This choice does not mean that use of external packages should be discouraged, just that their use or non-use should be thoroughly investigated and justified.
The code has been separated into modules in distinct python files, each containing related functions (BED functions, UI functions, exon coordinate functions and web API functions. They are imported into the main LRG_parser.py file and functions within them are invoked by referencing module.function_name(). This creates an uncluttered working environment, gives context to the use of particular functions, and modularity of code allows easy reuse in other projects. All functions perform single, distinct operations, meaning that tests can identify issues on a granular level.
Below is a diagram showing the modular design of the program.

Below is a link to a flowchart showing the operational flow of the program. It is expected that this can be used by future developers or maintainers. Program Flowchart
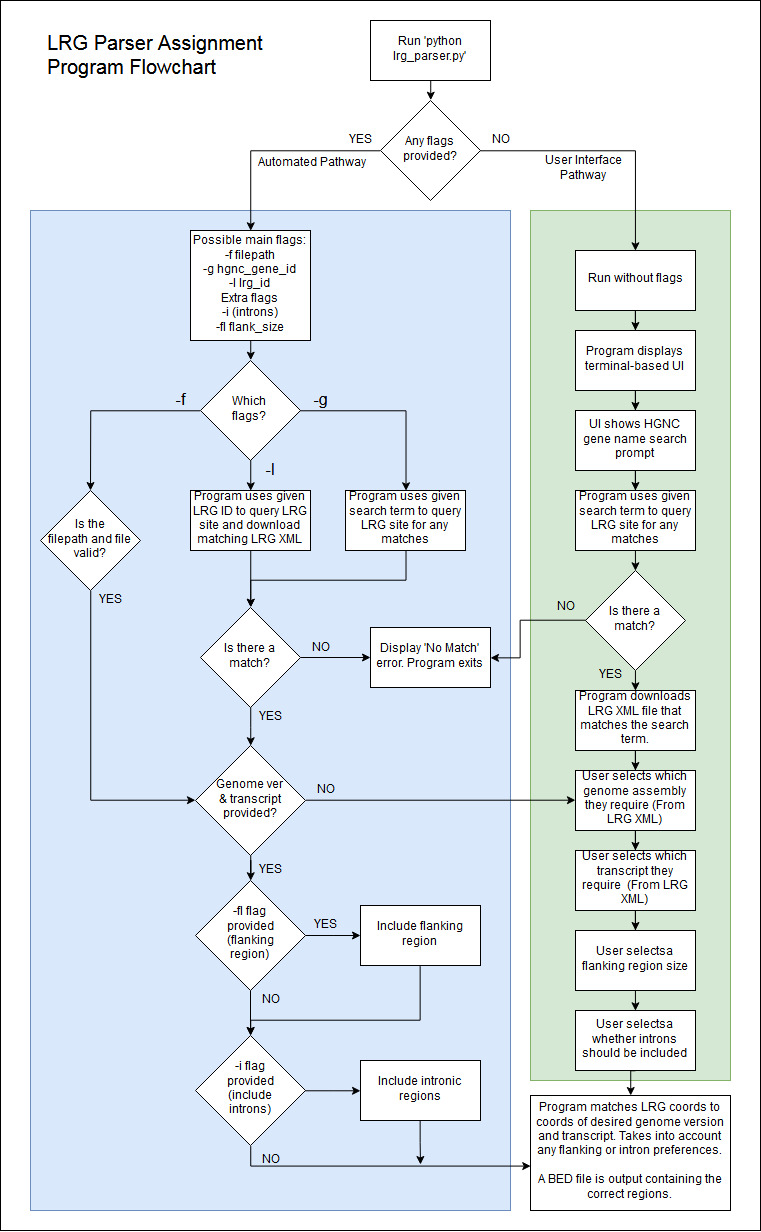{kind=link}
The Python library unittest has been used when writing tests. The test script is divided into separate classes for each module (ui.py, webservices.py, etc). Within each class, tests have been written for all of the functions within the specified module. This layout allows easy identification of the tests relating to each bit of code.
To check test coverage, the Python tool coverage.py (link) was used. This runs through the test.py file, and records the level of test coverage for the program. Below is the output for coverage.py. Test coverage levels for most of the modules written for the program are 100%.

Although not all of the lines of code have been tested, coverage.py identifies the lines which are not tested so that they can be reviewed more thoroughly.
Having a high level of test coverage is essential when developing software, as it allows accurate and efficient detection of any program-breaking changes. Changes in one part of the codebase may break or change how code works elsewhere so it is important to regularly run tests. Without tests, these unwanted changes may go unnoticed and lead to situations where inaccurate BED files are used, which is unacceptable in a clinical environment.
Tests can be run with the command:
python3 test.pyThe program can simply be installed by cloning the git repository.
git clone https://github.com/Addy81/lrg_project.git
There are no external dependencies or libraries used so no further installation steps are required.
The program can be used with or without flag arguments.
python lrgparser.pyAs no arguments have been provided, the program loads the UI and prompts the user for a gene, a desired genome version* and a desired transcript*.
*These are extracted from the LRG file which will be downloaded, no knowledge about available transcripts is required by the user.
The program is flexible and can take multiple optional flags as arguments.
Three flags are available to define the LRG to use. Only one of these should be provided:
| Short Flag | Long Flag | Description |
|---|---|---|
-f |
--file |
Takes an LRG XML file as an argument (e.g LRG_384.xml) |
-l |
--lrgid |
Takes an LRG ID as an argument (e.g LRG_384) |
-g |
--gene |
Takes an HGNC gene name as an argument (e.g MYH7) |
Additional flags can be used to indicate the desired reference genome version or the desired transcript. If these are not given, the UI will ask the user for their preference. If these are provided along with one of the flags from above, BED file generation will be completely automated.
| Short Flag | Long Flag | Description |
|---|---|---|
-t |
--transcript |
Takes a transcript as an argument (e.g NM_000257.2) |
-r |
--referencegenome |
Takes a reference genome as an argument (e.g GRCh37.p13) |
These are other optional arguments which are not required for automated BED generation, but they provide functionality that may be useful.
| Short Flag | Long Flag | Description |
|---|---|---|
-i |
--introns |
If this flag is present, intronic regions will be included |
-fl |
--flank |
Takes a flank size in bases (Minimum 0, Maximum 5000) |
-
When you know the gene name, but not the LRG ID and you don't have a file
python lrgparser.py -g MYH7
The UI will prompt for the user to choose a genome version and transcript. -
When you know the gene name, genome version and transcript you want
python lrgparser.py -g MYH7 -r GRCh37.p13 -t NM_000257.2
As all necessary arguments for fully automated BED file generation have been provided. The UI will not run and the BED file is created with no user input. -
When you have an LRG XML file, know the genome version but do not know which transcripts are available
python lrgparser.py -f LRG_384.xml -r GRCh37.p13
The program will look at the XML file to find available transcripts and display the UI to prompt the user to choose one. -
When you know the gene name and transcript, and you want a flanking region of 200 bases on each region
python lrgparser.py -g MYH7 -t NM_000257.2 -fl 200
The program will download the appropriate LRG XML file, parse it to find available genome versions and then display the UI to prompt the user to choose one. -
When you only know the LRG ID, but want the whole gene (both exonic and intronic regions)
python lrgparser.py -l LRG_384 -i
The program will download the appropriate LRG XML file, parse it to find available genome versions and transcripts. The UI will then prompt the user for their preference.