-
Notifications
You must be signed in to change notification settings - Fork 0
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
Showing
3 changed files
with
273 additions
and
10 deletions.
There are no files selected for viewing
163 changes: 163 additions & 0 deletions
163
assignments/lab/scrnaseq-bioconductor/assignment/index.md
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,163 @@ | ||
| # Single Cell RNA-seq Analysis with Bioconductor | ||
|
|
||
| ## Overview | ||
|
|
||
| During the lab session we practiced analyzing 10x Genomics data starting with output files from the 10x Genomics Cell Ranger pipeline. | ||
| While this demonstrates how you might analyze new data that you generate, there are many public datasets that you may wish to explore starting with the analysis already conducted by the original authors. | ||
| In this assignment, you will obtain data from the Fly Cell Atlas which characterized 15 tissue types alongside whole head and body. | ||
| Through a coordinated effort among >40 Drosophila labs around the world, more than 250 single-cell clusters were identified and annotated. | ||
| After loading and exploring the quality of the Gut dataset, you will identify marker genes to gain insights into biological functions. | ||
|
|
||
| Refer to the Bioconductor OSCA book for more information on | ||
|
|
||
| - https://bioconductor.org/books/OSCA.intro -- Loading data and working with SingleCellExperiment objects | ||
| - https://bioconductor.org/books/OSCA.basic -- Analyzing data from normalization to marker gene detection | ||
|
|
||
| Use `help()` to display the built-in R Documentation for a given function (e.g. `help(sort)`) after loading the appropriate package | ||
|
|
||
| 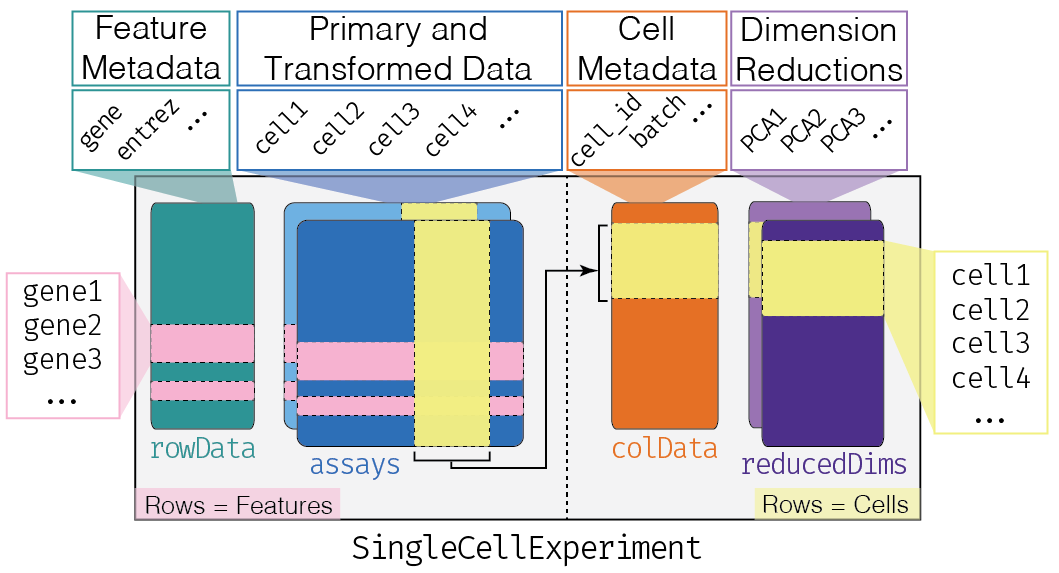 | ||
|
|
||
| ## Exercises | ||
|
|
||
| There are three exercises in this assignment: | ||
|
|
||
| 1. Load the Fly Cell Atlas Gut data and inspect the counts and cell metadata | ||
| 2. Explore the data at the gene-level, cell-level, and percent of mitochondrial reads | ||
| 3. Identify marker genes for subsets of epithelial cells and somatic precursor cells | ||
|
|
||
| Before you do anything else, create an R script for this assignment. | ||
| Everything you'll need to do for this assignment will be done in this script and submitted via GitHub. | ||
| Use comments to separate your code blocks into Exercise 1, Step 1.1; Exercise 1, Step 1.2; etc., and include written answers to questions as comments. | ||
| You must show your work to receive full credit for each question by providing the code you used. | ||
|
|
||
| ### 1. Load data | ||
|
|
||
| #### Load packages | ||
|
|
||
| Load the following packages using `library()` | ||
|
|
||
| - zellkonverter -- loads single cell data from the popular H5ad file format and creates a SingleCellExperiment object | ||
| - scuttle, scater, and scran -- provide core single cell functionality | ||
| - ggplot2 -- data visualization | ||
|
|
||
| #### Load and inspect data (0.5 pt) | ||
|
|
||
| Load Gut data from flycellatlas.org | ||
|
|
||
| - Download the Gut "10x, RAW, H5AD" data using a web browser (should be 612 MB) | ||
| - Create a SingleCellExperiment object named `gut` by loading `v2_fca_biohub_gut_10x_raw.h5ad` using `readH5AD()` from zellkonverter | ||
| - Change the assay name from `X` to `counts` using `assayNames(gut) <- "counts"` | ||
| - Normalize counts using `gut <- logNormCounts(gut)` | ||
|
|
||
| **Question 1**: Inspect the `gut` SingleCellExperiment object (0.5 pt) | ||
|
|
||
| - How many genes are quantitated (should be >10,000)? | ||
| - How many cells are in the dataset? | ||
| - What dimension reduction datasets are present? | ||
|
|
||
| #### Inspect cell metadata (0.5 pt) | ||
|
|
||
| **Question 2**: Inspect the available cell metadata (0.5 pt) | ||
|
|
||
| - How many columns are there in `colData(gut)`? | ||
| - Which three column names reported by `colnames()` seem most interesting? Briefly explain why. | ||
| - Plot cells according to `X_umap` using `plotReducedDim()` and colouring by `broad_annotation` | ||
|
|
||
| ### 2. Explore data | ||
|
|
||
| #### Explore gene-level statistics (1 pt) | ||
|
|
||
| Sum the expression of each gene across all cells | ||
|
|
||
| - Create a vector named `genecounts` by using `rowSums()` on the counts matrix returned by `assay(gut)` | ||
|
|
||
| **Question 3**: Explore the `genecounts` distribution (1 pt) | ||
|
|
||
| - What is the mean and median genecount according to `summary()`? What might you conclude from these numbers? | ||
| - What are the three genes with the highest expression after using `sort()`? What do they share in common? | ||
|
|
||
| #### Explore cell-level statistics (1 pt) | ||
|
|
||
| **Question 4a**: Explore the total expression in each cell across all genes (0.5 pt) | ||
|
|
||
| - Create a vector named `cellcounts` using `colSums()` | ||
| - Create a histogram of `cellcounts` using `hist()` | ||
| - What is the mean number of counts per cell? | ||
| - How would you interpret the cells with much higher total counts (>10,000)? | ||
|
|
||
| **Question 4b**: Explore the number of genes detected in each cell (0.5 pt) | ||
|
|
||
| - Create a vector named `celldetected` using `colSums()` but this time on `assay(gut)>0` | ||
| - Create a histogram of `celldetected` using `hist()` | ||
| - What is the mean number of genes detected per cell? | ||
| - What fraction of the total number of genes does this represent? | ||
|
|
||
| #### Explore mitochondrial reads (1 pt) | ||
|
|
||
| Sum the expression of all mitochondrial genes across each cell | ||
|
|
||
| - Create a vector named `mito` of mitochondrial gene names using `grep()` to search `rownames(gut)` for the pattern `^mt:` and setting `value` to TRUE | ||
| - Create a DataFrame named `df` using `perCellQCMetrics()` specifying that `subsets=list(Mito=mito)` | ||
| - Confirm that the mean sum and detected match your previous calculations by converting `df` to a data.frame using `as.data.frame()` and then running `summary()` | ||
| - Add metrics to cell metadata using `colnames(colData(gut)) <- cbind( colData(gut), df )` | ||
|
|
||
| **Question 5**: Visualize percent of reads from mitochondria (1 pt) | ||
|
|
||
| - Plot the `subsets_Mito_percent` on the y-axis against the `broad_annotation` on the x-axis rotating the x-axis labels using `theme( axis.text.x=element_text( angle=90 ) )` and **submit this plot** | ||
| - Which cell types may have a higher percentage of mitochondrial reads? Why might this be the case? | ||
|
|
||
| ### 3. Identify marker genes | ||
|
|
||
| #### Analyze epithelial cells (3 pt) | ||
|
|
||
| **Question 6a**: Subset cells annotated as "epithelial cell" (1 pt) | ||
|
|
||
| - Create an vector named `coi` that indicates cells of interest where `TRUE` and `FALSE` are determined by `colData(gut)$broad_annotation == "epithelial cell"` | ||
| - Create a new SingleCellExperiment object named `epi` by subsetting `gut` with `[,coi]` | ||
| - Plot `epi` according to `X_umap` and colour by `annotation` and **submit this plot** | ||
|
|
||
| Identify marker genes in the anterior midgut | ||
|
|
||
| - Create a list named `marker.info` that contains the pairwise comparisons between all annotation categories using `scoreMarkers( epi, colData(epi)$annotation )` | ||
| - Identify the top marker genes in the anterior midgut according to `mean.AUC` using the following code | ||
|
|
||
| ``` | ||
| chosen <- marker.info[["enterocyte of anterior adult midgut epithelium"]] | ||
| ordered <- chosen[order(chosen$mean.AUC, decreasing=TRUE),] | ||
| head(ordered[,1:4]) | ||
| ``` | ||
|
|
||
| **Question 6b**: Evaluate top marker genes (2 pt) | ||
|
|
||
| - What are the six top marker genes in the anterior midgut? Based on their functions at flybase.org, what macromolecule does this region of the gut appear to specialize in metabolizing? | ||
| - Plot the expression of the top marker gene across cell types using `plotExpression()` and specifying the gene name as the feature and `annotation` as the x-axis and **submit this plot** | ||
|
|
||
| #### Analyze somatic precursor cells (3 pt) | ||
|
|
||
| Repeat the analysis for somatic precursor cells | ||
|
|
||
| - Subset cells with the broad annotation `somatic precursor cell` | ||
| - Identify marker genes for `intestinal stem cell` | ||
|
|
||
| **Question 7**: Evaluate top marker genes (3 pt) | ||
|
|
||
| - Create a vector `goi` that contains the names of the top six genes of interest by `rownames(ordered)[1:6]` | ||
| - Plot the expression of the top six marker genes across cell types using `plotExpression()` and specifying the `goi` vector as the features and **submit this plot** | ||
| - Which two cell types have more similar expression based on these markers? | ||
| - Which marker looks most specific for intestinal stem cells? | ||
|
|
||
| ## Submit | ||
|
|
||
| Submit an R script with your code and answers to questions for | ||
|
|
||
| - Loading data (1 pt) | ||
| - Exploring data (2.5 pt) | ||
| - Identifying marker genes (3 pt) | ||
|
|
||
| Be sure to include the following plots | ||
|
|
||
| - Broad annotation vs % mitochondrial reads (0.5 pt) | ||
| - Epithelial cells UMAP colored by annotation (1 pt) | ||
| - Expression plot of top marker gene in epithelial cells according to annotation (1 pt) | ||
| - Expression plot of top six marker genes in precursor cells according to annotation (1 pt) | ||
|
|
101 changes: 101 additions & 0 deletions
101
...nments/lab/scrnaseq-bioconductor/slides_asynchronous_or_livecoding_resources/code-along.R
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,101 @@ | ||
| # | ||
| # Basic single cell analysis inspired by Bioconductor OSCA Quick start | ||
| # - https://bioconductor.org/books/3.19/OSCA.intro/analysis-overview.html#quick-start-simple | ||
| # | ||
|
|
||
|
|
||
|
|
||
| ### Load packages | ||
|
|
||
| library( "DropletUtils" ) # Utilities for handling 10x Genomics data | ||
| library( "scater" ) # Tools for quality control and visualization | ||
| library( "scran" ) # Methods for single cell data analysis | ||
|
|
||
|
|
||
|
|
||
| ### Load data | ||
|
|
||
| # wget https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz | ||
| sce <- read10xCounts( "filtered_gene_bc_matrices/hg19/" ) | ||
| sce | ||
|
|
||
|
|
||
|
|
||
| ### Explore SingleCellExperiment | ||
|
|
||
| # cell metadata | ||
| colData(sce) | ||
| # convert from DataFrame to data.frame | ||
| as.data.frame(colData(sce)) | ||
|
|
||
| # gene metadata | ||
| rowData(sce) | ||
| # change from ID to Symbol | ||
| rownames(sce) <- rowData(sce)$Symbol | ||
| sce | ||
|
|
||
| # subset matrix and SingleCellExperiment with [row,col] | ||
| assay(sce)[40:50,1:10] | ||
| assay(sce[40:50,1:10]) | ||
| assay(sce[40:50,])[,1:10] | ||
|
|
||
| # sum up rows or columns for insight | ||
| genecounts <- rowSums(assay(sce)) | ||
| summary( genecounts ) | ||
| head( sort(genecounts, decreasing=TRUE ) ) | ||
|
|
||
|
|
||
|
|
||
| ### Basic processing | ||
|
|
||
| # Library size normalization and log-transformation | ||
| sce <- logNormCounts( sce ) | ||
|
|
||
| # Quantify per-gene variation | ||
| dec <- modelGeneVar(sce) | ||
| # Select highly variable genes | ||
| hvg <- getTopHVGs(dec, prop=0.1) | ||
|
|
||
| # Principal component analysis | ||
| set.seed(1234) # Set seed for reproducibility | ||
| sce <- runPCA( sce, subset_row=hvg ) | ||
| plotReducedDim( sce, "PCA" ) | ||
|
|
||
|
|
||
|
|
||
| ### Clustering | ||
|
|
||
| # Principal components are used for clustering | ||
| colLabels(sce) <- clusterCells(sce, use.dimred='PCA') | ||
| as.data.frame(colData(sce)) | ||
| plotReducedDim( sce, "PCA", colour_by="label" ) | ||
|
|
||
|
|
||
|
|
||
| ### Additional visualizations | ||
|
|
||
| # TSNE and UMAP primarily for visualization | ||
| set.seed(1234) # Set seed for reproducibility | ||
| sce <- runTSNE(sce, dimred = 'PCA') | ||
| plotReducedDim( sce, "TSNE", colour_by="label" ) | ||
|
|
||
| # Plotting distributions is a good complement | ||
| rownames(sce) <- rowData(sce)$Symbol | ||
| goi <- c( "MS4A1", "CD14", "CD8A", "IL7R" ) | ||
| plotExpression( sce, features=goi, x="label" ) | ||
|
|
||
|
|
||
|
|
||
| ### Finding marker genes | ||
|
|
||
| # All-vs-all comparison of clusters | ||
| marker.info <- scoreMarkers(sce, colData(sce)$label) | ||
| marker.info | ||
|
|
||
| # Sort based on AUC | ||
| chosen <- marker.info[["11"]] | ||
| ordered <- chosen[order(chosen$mean.AUC, decreasing=TRUE),] | ||
| head(ordered[,1:4]) | ||
|
|
||
| plotReducedDim( sce, "TSNE", colour_by="CD79A" ) | ||
| plotExpression( sce, "CD79A", x="label" ) |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -1,19 +1,18 @@ | ||
| # QuantLab Week 8 - The 3D Genome | ||
| # QuantLab Week 8 - # Single Cell RNA-seq Analysis with Bioconductor | ||
|
|
||
| Assignment Date: Friday, Nov. 10, 2023 | ||
| Assignment Date: Friday, Nov. 1, 2024 | ||
|
|
||
| Due Date: Friday, Nov. 17, 2023 | ||
| Due Date: Friday, Nov. 8, 2024 | ||
|
|
||
| ## Lecture -- Michael Sauria | ||
| ## Code Along | ||
|
|
||
| [Lecture Slides](https://www.dropbox.com/scl/fi/0cek4f6vr6ziw4l72mmr0/3D-Genome-2023.pdf?rlkey=gsqbsosw1cu339vdtwom86552&dl=0) | ||
| [Slidedeck](https://docs.google.com/presentation/d/1NiE1wPoxLMGImj_sHkuFfOeNyaqdvsOH5lIdOPRx9i8) | ||
|
|
||
| ## Live coding resources | ||
|
|
||
| You will need to erase old versions of the conda install scripts from last week (`rm ~/conda_install*`). The new versions of the scripts can be found at [github](https://www.github.com/bxlab/cmdb-laptop-setup), [conda_install_arm64.sh](https://github.com/bxlab/cmdb-laptop-setup/raw/master/conda_install_arm64.sh) and [conda_install_x86.sh](https://github.com/bxlab/cmdb-laptop-setup/raw/master/conda_install_x86.sh). | ||
| The code along script can be found [here](https://github.com/bxlab/cmdb-quantbio/blob/main/assignments/lab/scrnaseq-bioconductor/slides_asynchronous_or_livecoding_resources/code-along.R) | ||
|
|
||
| ## Homework Assignment | ||
|
|
||
| Complete the homework assignment in your `week8` submission directory in your `qbb2023-answers`. | ||
| Complete the homework assignment in your `week8` submission directory in your `qbb2024-answers`. | ||
|
|
||
| [Homework Assignment](../assignments/lab/scrnaseq-bioconductor/assignment/) | ||
|
|
||
| [Homework Assignment](../assignments/lab/3D_genome/assignment/) |