-
Notifications
You must be signed in to change notification settings - Fork 28
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
Showing
1 changed file
with
208 additions
and
0 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,208 @@ | ||
| --- | ||
| date: 2023-10-14 | ||
| comments: true | ||
| --- | ||
| # 生信爱好者周刊(第 95 期):中国人群泛基因组联盟 | ||
|
|
||
| 这里记录每周值得分享的生信相关内容,周日发布。 | ||
|
|
||
| 本杂志开源(GitHub: [openbiox/weekly](https://github.com/openbiox/weekly "openbiox/weekly")),欢迎提交 issue,投稿或推荐生信相关内容。 | ||
|
|
||
| [「生信周刊讨论区」](https://github.com/openbiox/weekly/discussions "「生信周刊讨论区」") | ||
|
|
||
| ## 封面图 | ||
|
|
||
|  | ||
|
|
||
| ## 本周话题:[中国人群泛基因组联盟](https://mp.weixin.qq.com/s/0MJ_CBJbdPj6VZTOMUMdLg) | ||
| > 中国人群泛基因组联盟是由复旦大学、西安交通大学、中国医学科学院等26家单位发起的一个科研项目,旨在构建能够代表中华民族遗传多样性的中国人群泛基因组图谱。 | ||
| > | ||
| > 目前,通用的人类参考基因组主要是基于欧洲白人为主体样本构建的,难以代表非欧裔族群、尤其是我国族群的基因组多样性。而我国作为人口大国,拥有丰富的人群多样性和悠久的人群历史,其遗传特征对于理解人类起源与演化、疾病易感性与表型多样性、精准医学与健康中国战略都具有重要意义。 | ||
| > | ||
| > 因此,建立我国自己的人群泛基因组图谱十分必要和紧迫。这也是中国人群泛基因组联盟的主要目标之一。在其第一期研究中,CPC对代表中国36个族群的58个样本进行了深度测序和高质量单倍型基因组组装,构建了高质量中国人群参考泛基因组。 | ||
| @NiEntropy :泛基因组与我们熟知的参考基因组的区别,参考基因组是一个线性的、单一的、一维的基因组序列,通常是从一个或少数几个个体中选取的代表性样本,不能完全反映个体之间的遗传变异。泛基因组是一个图形的、多样的、多维的基因组序列集合,包含了一个物种或一个群体中所有个体的遗传变异信息,能更好地反映基因组的结构和功能。参考基因组是泛基因组的一个子集,泛基因组比参考基因组包含了更多的新序列、新变异和新功能。 | ||
|
|
||
| @eplants:[为什么要开展泛基因组测序?](https://zhuanlan.zhihu.com/p/176135716)在漫长的进化过程中,由于地域因素,环境因素等的影响,每个个体都形成了极其特别的遗传性状,单一个体的基因组已经不能涵盖这个物种的所有遗传信息。 | ||
|
|
||
| ## 生信研究 | ||
| 1、[Science | 错义突变预测模型AlphaMissense](https://mp.weixin.qq.com/s/cLxyItI35oFJ36u-wDZ6Qw) | ||
|
|
||
|  | ||
|
|
||
| AlphaMissense是一种能够识别DNA中导致蛋白质氨基酸变化的错义突变的人工智能模型。它基于AlphaFold模型,结合了大量的遗传和群体数据,可以预测71百万个错义突变的致病性或良性。它的准确率高达89%,远超人类专家的水平。它旨在帮助快速诊断和治疗遗传疾病,并向研究社区开放了预测结果和模型代码。 | ||
|
|
||
| - 论文链接:https://www.science.org/doi/10.1126/science.adg7492 | ||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
| 2、[Cancer Cell|基于AI 的生物标志物预测模型加速CRC活检分析](https://mp.weixin.qq.com/s/_9fNnenKZUInEeP3rSSQTA) | ||
|
|
||
|  | ||
|
|
||
| 研究团队开发了一种新的基于Transformer的分析框架,通过将预训练的Transformer编码器与用于补丁聚合的Transformer网络相结合,从CRC常规病理切片中进行端到端的生物标志物预测。通过对来自16个队列13,000多名结直肠癌患者组成的大规模、多中心队列进行训练和评估,该方法在手术切除组织样本的MSI预测方面达到了99%的灵敏度,阴性预测值超99%。与当前最先进的算法相比,基于Transformer的方法大大提高了预测性能、通用性、数据效率和可解释性。 | ||
|
|
||
| - 论文链接:https://www.cell.com/cancer-cell/pdf/S1535-6108(23)00278-7.pdf | ||
|
|
||
|
|
||
|
|
||
| 3、[ Genome Biology | 用于单细胞RNA-seq和ATAC-seq数据多视图分析的深度生成模型 ](https://mp.weixin.qq.com/s/SceVchGE7G6pHsbUaBPRLQ) | ||
| 本文作者提出了一种多模态深度生成模型——单细胞多视图分析器(scMVP),它被设计用于处理同时测量同一细胞中基因表达和染色质可及性的测序数据,包括来自10X Genomics的SNARE-seq,sci-CAR,Paired-seq,SHARE-seq和Multiome方法。scMVP通过聚类一致性约束的多视图变分自编码器模型(VAE)自动学习scRNA-seq和scATAC-seq数据的共同潜在表示,并通过分层数据生成过程,包括变压器基于自关注的scATAC生成通道和基于掩码关注的scRNA生成通道,从多组数据的共同潜在嵌入中推导出各单层数据。scMVP可以帮助缓解数据稀疏问题,并通过共同的潜在嵌入准确识别不同联合分析技术的细胞群。最后,作者在几个实际数据集上展示了scMVP的优势。 | ||
| 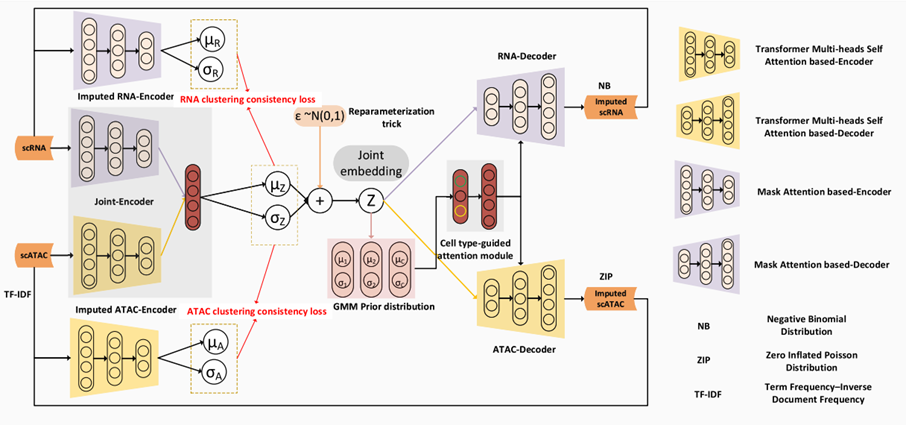 | ||
|
|
||
| - 论文链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02595-6 | ||
|
|
||
|
|
||
|
|
||
| ## 博文资讯 | ||
| 4、[ 线性模型中的高级特征选择技术(岭回归,LASSO,弹性网络)](https://mp.weixin.qq.com/s/aG1ooRC0jiVvrV44-xnIyw) | ||
|
|
||
| 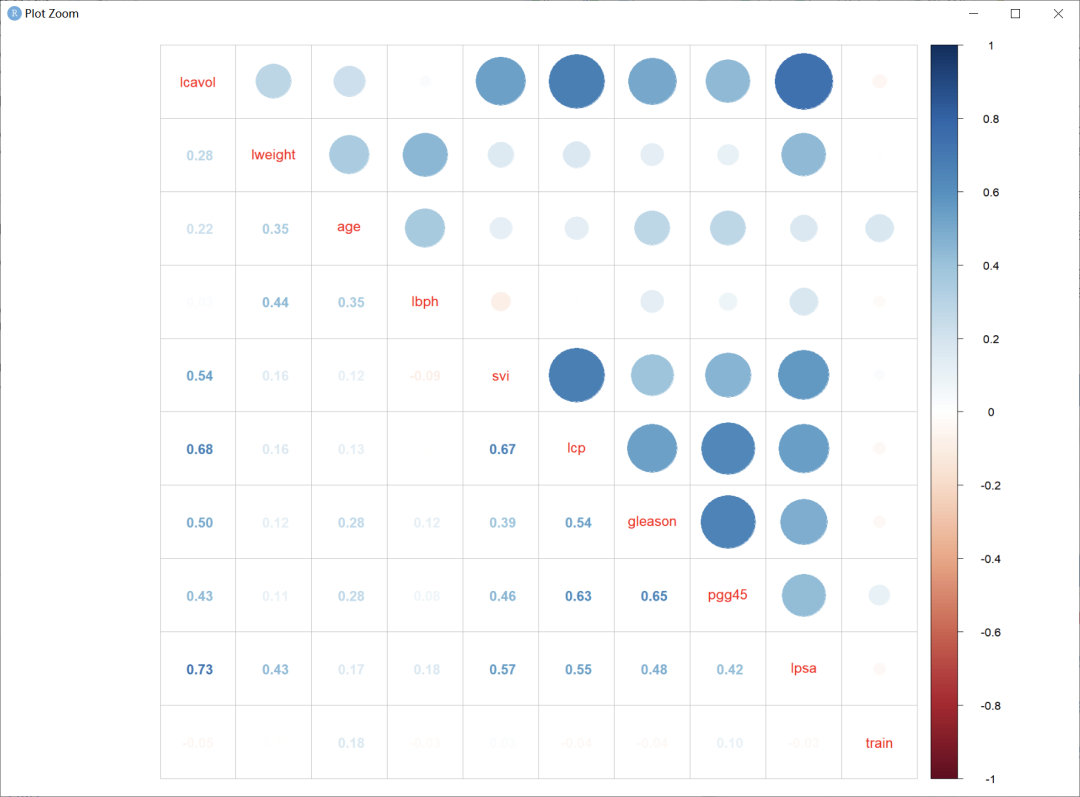 | ||
|
|
||
| 线性模型在机器学习问题中是非常有效的。当今很多数据集具有数量庞大的特征(feature),即高维性。此外,随着我们要处理的数据规模不断增大,最优子集和逐步特征选择这样的技术会造成难以承受的时间成本。在很多情况下,要得到一个最优子集的解需要花费数小时。 | ||
| 这种情况下有更好的解决方式。本文会讨论正则化的概念,正则化会对系数进行限制,甚至将其缩减到0。现在有很多种方法和方法组合可以实现正则化,我们会集中讨论岭回归和最小化绝对收缩和选择算子,最后讨论弹性网络,它将前面两种算法的优点合二为一。 | ||
|
|
||
|
|
||
|
|
||
| 5、[GREAT富集分析时请使用正确的基因组版本](https://mp.weixin.qq.com/s/ihIOZNXc9QcJc2JouPyzFg) | ||
|
|
||
| 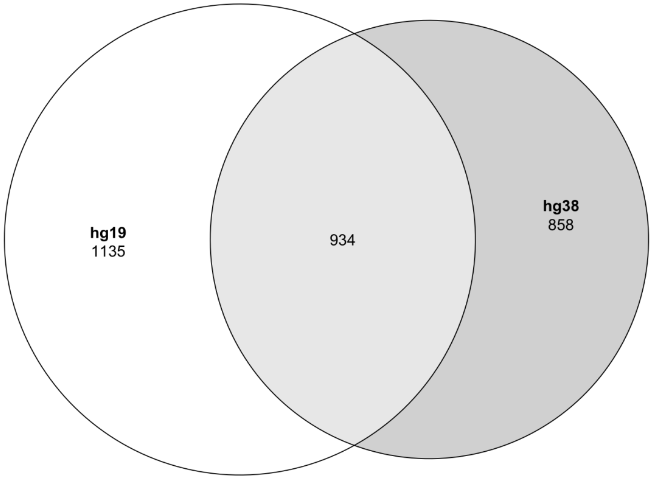 | ||
|
|
||
|
|
||
| GREAT分析是用来直接对基因组区间进行功能富集的一种分析方法,这里通过一个例子说明分错误的基因组版本会对结果产生非常大的影响,尤其是那些只关联了较少的用户基因组区间的基因集合。因此在进行GREAT分析时,选择正确的基因组版本非常重要。 | ||
|
|
||
| 6、[小牛生信 | R 语言基础课总结笔记](https://mp.weixin.qq.com/s/kKhmsczVrpuJDN-OGqnTpg) | ||
|
|
||
|  | ||
|
|
||
| 这篇推文是一位生物信息学爱好者写的R语言基础课总结笔记。内容包括: | ||
|
|
||
| - R语言的预备知识,如获取和设置工作目录,变量名的命名,赋值,注释,查看和清除变量等。 | ||
| - R语言的数据类型和数据结构,如向量,矩阵,数据框,列表等,以及如何生成,赋值,取子集,修改,合并,排序等操作。 | ||
| - R语言的函数和参数,如如何使用内置函数,查看帮助文档,自定义函数等。 | ||
| - R语言的R包,如如何安装和调用R包,以及一些常用的R包的介绍和使用方法。 | ||
| - R语言的文件读写,如如何读取和导出不同格式的文件,以及R特有的数据格式Rdata等。 | ||
|
|
||
|
|
||
|
|
||
| 7、[ Oxford Nanopore 在London Calling大会上宣布突破性的技术性能和路径,让任何人可以在任何地方测序](https://mp.weixin.qq.com/s/BVhdGextw_tLpEdwlFMvJw) | ||
|
|
||
|  | ||
|
|
||
| 在2023年5月17日至19日举行的公司年度London Calling大会上,Oxford Nanopore的首席技术、创新和产品官Clive G Brown在技术更新介绍中宣布了突破性的性能数据和新平台的最新情况,包括直接RNA测序以支持以RNA为基础的疗法的发展。 | ||
| 随着Oxford Nanopore社区用户群体不断扩大,覆盖的科研领域也更加广泛,这些改进的工具正在临床研究和工业应用市场中建立起应用场景,这些市场需要近样品、可访问的实时分析工具。而之前,由于可及性和效用性等障碍,测序还未被广泛应用于这些场景中。 | ||
|
|
||
|
|
||
|
|
||
| ## 工具 | ||
| 8、[funkyheatmap | 显示混合数据类型的数据框](https://github.com/funkyheatmap/funkyheatmap) | ||
|
|
||
|  | ||
|
|
||
| 9、[grf | 广义随机森林R包](https://github.com/grf-labs/grf) | ||
|
|
||
|
|
||
|
|
||
|  | ||
|
|
||
| 斯坦福大学和其他机构的研究人员开发和维护的名为**grf**的R语言包,它可以实现**广义随机森林**(Generalized Random Forests)的方法。广义随机森林是一种基于树的统计估计和推断的非参数方法。 | ||
|
|
||
| - 教程链接:https://grf-labs.github.io/grf/ | ||
|
|
||
|
|
||
|
|
||
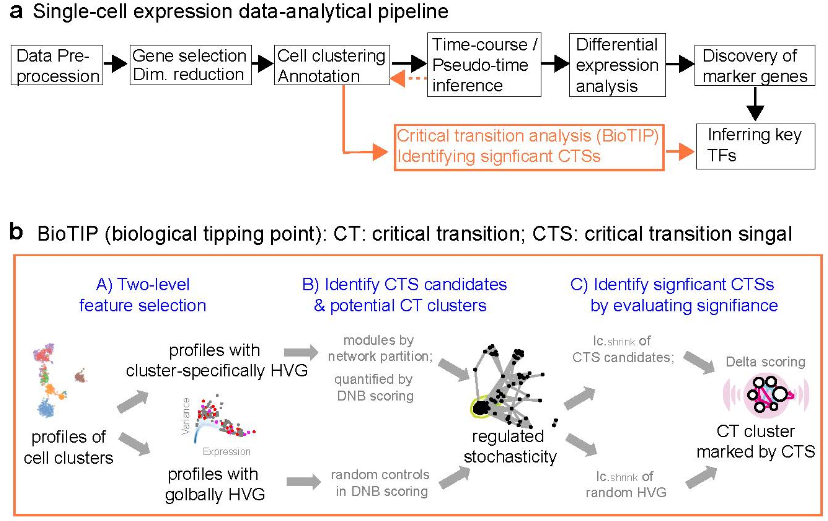
| 10、[BioTIP:用于表征生物临界点的 R 包](https://github.com/openbiox/weekly/issues/1684) | ||
|
|
||
|  | ||
|
|
||
|
|
||
| 突然且通常不可逆转的变化(或临界点)在生物过程的进展中具有决定性作用。这个 R 包可以用于使用基因表达谱来表征生物临界点。 BioTIP 具有两种功能:(1) 准确检测临界点,(2) 识别重要的关键转变信号 (CTS)。 | ||
|
|
||
|
|
||
|
|
||
| 11、[NanoBlot | 用于从长读 RNA 测序数据中可视化 RNA 同工型的 R 软件包](https://github.com/SamDeMario-lab/NanoBlot) | ||
|
|
||
|  | ||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
| 12、[ggside](https://github.com/jtlandis/ggside) | ||
|
|
||
|
|
||
|
|
||
|  | ||
|
|
||
| 这个工具是一个名为ggside的R语言包,它可以扩展ggplot2包的功能。它可以让用户在主图的某个轴上添加图形信息,比如离散轴的元数据,或者连续轴的汇总图形,如箱线图或密度分布。 | ||
|
|
||
|
|
||
|
|
||
| ## 资源 | ||
|
|
||
|
|
||
| 13、[丁鹏在伯克利的因果推断讲义](https://mp.weixin.qq.com/s/BtjOnHnJbQR9NYk3ELADEQ) | ||
|
|
||
|  | ||
|
|
||
|
|
||
|
|
||
| 一本关于因果推断的新书A First Course in Causal Inference,地址:https://arxiv.org/abs/2305.18793 本书中文版正由作者丁鹏亲自翻译,名曰《因果推断基础教程》,预计不久后将正式出版。 | ||
|
|
||
| 本书是根据丁鹏老师过去七年里,在加州大学伯克利分校的“因果推断”课程的基础上编写的。受众都是本科生和研究生,该讲义只需要概率论、统计推断以及线性回归、逻辑回归的基础知识。如果您发现任何错误,请随时给作者发电子邮件。 | ||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
| 14、[LangChain and LlamaIndex Projects Lab Book](https://leanpub.com/langchain/read) | ||
|
|
||
|  | ||
|
|
||
| 一本使用python语言来介绍 LangChain 和 LlamaIndex 项目, 通过使用 LangChain 和 LlamaIndex,结合开放AI的模型来解决一系列有趣的问题,比如信息检索、自然语言处理等。值得有兴趣的的同学翻阅。 | ||
|
|
||
|
|
||
|
|
||
| 15、[Human Genome Variation Lab](https://mccoy-lab.github.io/hgv_modules/authors.html) | ||
|
|
||
|  | ||
|
|
||
| 由The Johns Hopkins Data Science Lab 提供的人类基因组变异与计算课程,帮助学生学习人类基因组变异的原理和方法,以及如何使用计算工具进行分析和解释。 | ||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
| ## 历史上的本周 | ||
| - 第55期:[科学创新四十年,我们可能还没搞明白科学和技术的基本概念](https://mp.weixin.qq.com/s/QX9lowPT1b64hJUL1ZMXBA) | ||
|
|
||
| ## 贡献者(GitHub ID) | ||
|
|
||
| 「Openbiox 生信周刊」运维小队: | ||
|
|
||
| - `@ShixiangWang`(王诗翔) | ||
| - `@kkjtmac`(阚科佳) | ||
| - `@NiEntropy`(赵启祥) | ||
| - `@He-Kai-fly`(何凯) | ||
| - `@JnanZhang`(张佳楠) | ||
| - `@Tomcxf`(陈啸枫) | ||
| - `@wangdepin`(王德品) | ||
| - `@kongjianyang`(空间阳) | ||
|
|
||
| ## 订阅 | ||
|
|
||
| 这个周刊每周日发布,同步更新在微信公众号「优雅R」(elegant-r)上。 | ||
|
|
||
| 微信搜索“优雅R”或者扫描二维码,即可订阅。 | ||
|
|
||
|  | ||
|
|
||
| (完) |