A JBrowse plugin for viewing multiple alignments
Convert the MAF into a pseudo-BED format by calling bin/maf2bed.pl
bin/maf2bed.pl hg38 < file.maf > output.txt
bgzip output.txt
tabix -p bed output.txt.gz
The second argument to maf2bed.pl is the genome version e.g. hg38 used for the main species in the MAF (if your MAF comes from a pipeline like Ensembl or UCSC, the identifiers in the MAF file will say something like hg38.chr1, therefore, the argument to maf2bed.pl should just be hg38 to remove hg38 part of the identifier. if your MAF file does not include the species name as part of the identifier, you should add the species into them the those scaffold/chromosome e.g. create hg38.chr1 if it was just chr1 before)
If all is well, your BED file should have 6 columns, with chr, start, end, id, score, alignment_data, where alignment_data is separated between each species by ; and each field in the alignment is separated by :.
Note: you can also stream from a gzipped MAF to the bgzipped bed
gunzip -c chr21.maf.gz | bin/maf2bed.pl hg38 | bgzip > output.txt.gz
The bin/convert.sh script has a small automatic processing from maf to bgzipped, tabixed, bed.
- samples - an array of species in the MAF file (e.g. hg38, mm10, etc.)
- labelWidth - an integer width for labels (default: 100)
- style.matchColor - color to use for matches (default: green)
- style.mismatchColor - color to use for mismatches (default: blue)
- style.gapColor - color to use for gaps in alignment (default: red)
- style.mismatchBases - set to true, then you can set style.mismatchA, style.mismatchG, style.mismatchC, style.mismatchT as needed
Note: samples can be the array of strings like ["hg38","mm10"] or an array of extended JSON structures in order to customize the subtrack labels e.g. {"id": "hg38", "label": "Human", "description": "Extended description of species", "color": "rgb(255,255,0)" }
{
"label": "MAF",
"urlTemplate": "chrI.txt.gz",
"storeClass": "MAFViewer/Store/SeqFeature/MAF",
"type": "MAFViewer/View/Track/MAF",
"samples": [
"cb4",
"caeRem4",
"caePb3",
"caeSp111",
"caeJap4"
]
}
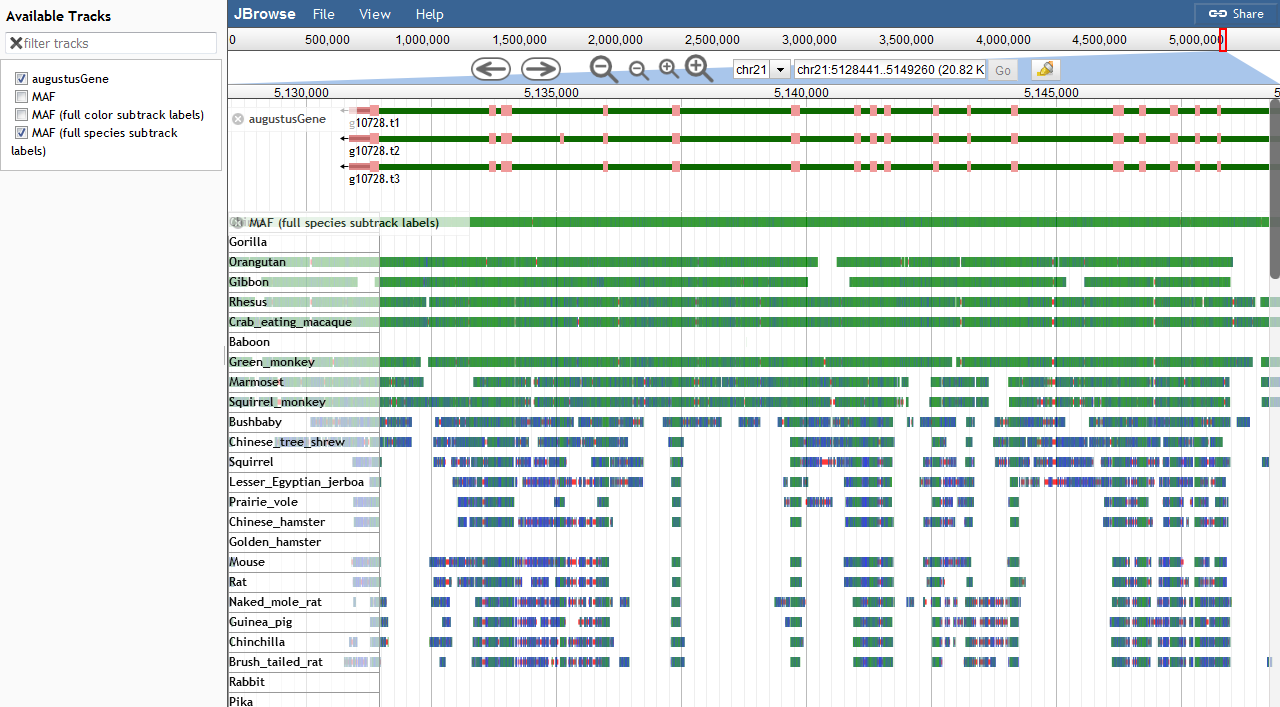
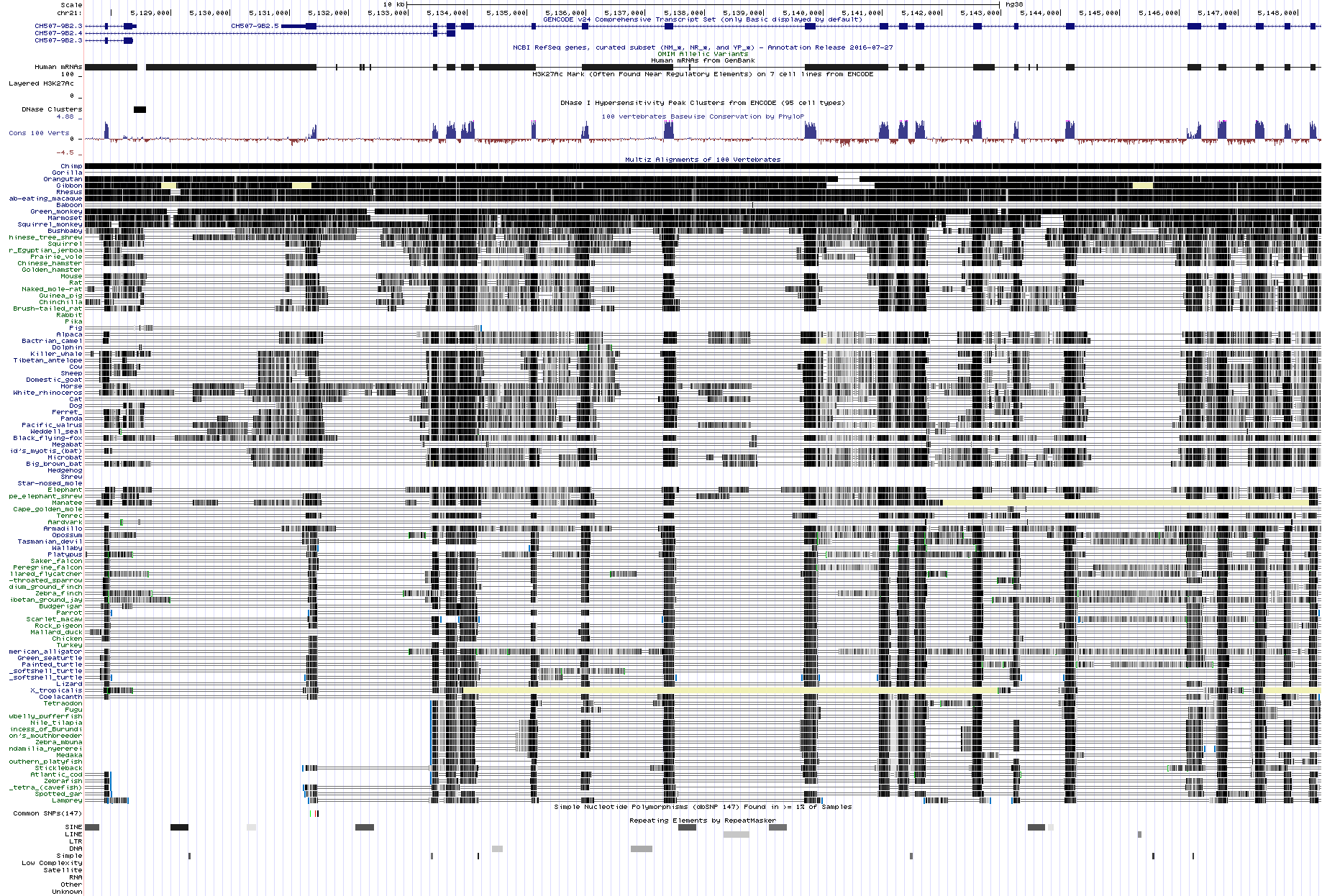
For comparison this is the same region in UCSC browser (picture) (link)
{kind=link}
Download to the plugins/MAFViewer and add to your config file with
"plugins": ["MAFViewer"]
See JBrowse FAQ on installing plugins
The test/ directory contains sample data for C. Elegans (from UCSC), Human (from UCSC), Medaka (from Ensembl).
Visit http://localhost/jbrowse/?data=plugins/MAFViewer/test/data or http://localhost/jbrowse/?data=plugins/MAFViewer/test/medaka or http://localhost/jbrowse/?data=plugins/MAFViewer/test/hg38 to view
You can obtain MAF output from lastz using --format=maf and then use mafviewer to compare two different genomes
You can also obtain MAF from mummer by converting the outputted .delta file to MAF with the delta2maf program (not distributed in the latest mummer versions, you can obtain delta2maf by downloading mugsy from sourceforge and finding delta2maf inside their version of mummer. Then run delta2maf yourfile.delta > yourfile.maf note that delta2maf assumes the fasta files are located where the .delta line 1 says they are)
Important: if using MAF files from lastz or mummer here, you should edit the MAF to include the organisms name pre-pended onto the chromosome names, e.g. if it says chr1, add "human.chr1" where relevant. Then the bin/maf2bed.pl program included in this package can be run with bin/maf2bed.pl human < yourfile.maf > output.bed which then strips the "human" part of the chromosome identifiers again
Requires JBrowse 1.12.3 or later for BEDTabix functionality
Feel free to provide feedback!