For the full documentation and user guide to ThermoPack, see the ThermoPack homepage.
If you are running ThermoPack installed via pip, make sure to check the documentation for the correct version by selecting
the version number in the sidebar.
Thermopack is a thermodynamics library for multi-component and multi-phase thermodynamics developed at SINTEF Energy Research and NTNU Department of Chemistry. Through decades of research, we have developed a software that performs thermodynamic calculations. A large selection of equations of state has been implemented in this software. Most of these equations of state have been developed by other research groups around the world, but some of them have been developed by us. Thermopack has been a much-appreciated in-house powerhouse.
Thermopack is available for everybody, free of charge under the MIT/Apache 2.0 open-source licenses. Thermopack is written in FORTRAN to handle heavy numerical computations associated with process and computational fluid dynamics (CFD) simulations. The thermodynamic framework is easily interfaced from C/C++ and also contains a flexible Python wrapper to make scripting easy.
- Please cite
- Authors and contact persons
- License
- Acknowledgments
- Program structure
- Building from Source
- Getting started - Python
- Doing calculations
- Advanced usage - Python
- Component identifiers
Thermopack has been developed through many projects, and have produced many articles. If you are writing academic publications, please cite one or more of the following articles:
-
For general usage:
Thermodynamic Modeling with Equations of State: Present Challenges with Established Methods -
Quantum cubic:
Accurate quantum-corrected cubic equations of state for helium, neon, hydrogen, deuterium and their mixtures -
SAFT-VR Mie and SAFT-VRQ Mie:
Equation of state and force fields for Feynman--Hibbs-corrected Mie fluids. I. Application to pure helium, neon, hydrogen, and deuterium
Equation of state and force fields for Feynman–Hibbs-corrected Mie fluids. II. Application to mixtures of helium, neon, hydrogen, and deuterium
Choice of reference, the influence of non-additivity and challenges in thermodynamic perturbation theory for mixtures -
CPA, PC-SAFT or cubic models with Wong–Sandler, Huron–Vidal or UNIFAC mixing rules:
Thermodynamic models to accurately describe the PVTxy-behavior of water/carbon dioxide mixtures -
Using dry-ice and water-ice model or the tc-PR/tc-RK:
Depressurization of CO2-N2 and CO2-He in a pipe: Experiments and modelling of pressure and temperature dynamics -
Energy-density and entropy-density flashes:
The influence of CO2 mixture composition and equations of state on simulations of transient pipeline decompression -
Mapping spinodals or critical points:
The spinodal of single-and multi-component fluids and its role in the development of modern equations of state
Predicting triggering and consequence of delayed LNG RPT -
Perturbation theories for Lennard-Jones spline fluid:
Perturbation theories for fluids with short-ranged attractive forces: A case study of the Lennard-Jones spline fluid
Thermodynamic properties of the 3D Lennard-Jones/spline model
Thermopack is distributed under the MIT license and Apache 2.0.
A number of colleagues at SINTEF Energy Research and NTNU have contributed to the development of thermopack. We gratefully acknowledge their contributions.
Each EoS in thermopack is a class, which inherits from the thermopack class found in thermo.py. The primary documentation
for the thermopack python wrapper consists of the docstrings of the thermopack class.
This class contains all generic methods used to compute thermodynamic properties, phase equilibria, etc. The inheriting
classes simply ensure that the correct part of the Fortran-module is linked when performing calculations, and provide
some extended functionality for handling EoS parameters and such. See the wiki
for more information on this.
Fluid parameters are compiled into the Fortran-module, and are not directly accessible through the Python-wrapper.
The entire fluid parameter database used by thermopack may be found in the /fluids directory
in the GitHub repo. In order to model fluids not currently supported in the module available through pip, thermopack
must be compiled from source with the new parameters. See the wiki
for information on how to add new fluids, and the GitHub README for a guide
on how to compile from source. Please feel free to leave a PR for new parameter sets such that these can be included in
future releases of thermopack.
Brief description of file structure:
thermopack/: Main library folder containing make scripts etc.thermopack/src/: Main path for Fortran source codethermopack/unittest/: Test files written for pFUunitthermopack/bin/: Compiled binaries and librariesthermopack/doc/: Memos and doxygen documentationthermopack/fluids/: Pure fluid filesthermopack/binaries/: Files containing binary interaction parameters etc.thermopack/MSVStudio/: Microsoft Visual Studio project and solution filesthermopack/include/: C/C++ include filethermopack/pyplot/: Example plot-scripts (Plotting text files generated by Thermopack)thermopack/addon/: Add-on functionalitythermopack/addon/pycThermopack/: Python interfacethermopack/addon/pyUtils/: Python utilities for generating fortran code and makefile input.thermopack/addon/trend_interface/: Interface for working with the TREND/EOSCG library developed by Roland Span and Ruhr-Universität Bochum
Thermopack has been compiled for Windows, Linux and macOS and made available on the Python Package Index (pypi), and can be installed using pip
pip3 install thermopackFor documentation on the version available on pypi, refer to the appropriate version number in the sidebar.
Pre-built wheels for the latest version of ThermoPack on GitHub are available for download here. Refer to the linked page for instructions on how to install packages directly from a python wheel. Please note that the latest version on GitHub may be less stable, tested, and well documented than the versions distributed on PyPI.
The following sections show how to fetch, compile and install Thermopack and the Python frontend pycThermopack. When things are properly installed, it may be useful to look into the examples provided in the getting started guide, and the pyExamples.
Thermopack source code can be compiled with the GNU Fortran compiler or Intel FORTRAN and is dependent on the LAPACK and BLAS libraries. On the Windows OS the code can be compiled using Microsoft Visual Studio. A solution file is found in thermopack/MSVStudio, assuming that the Intel Fortran compiler is integrated with Microsoft Visual Studio.
For macOS and Linux, Lapack and Blas can likely be installed using apt, brew, or similar. For windows, Lapack will need to be built from source. The CMake setup for Windows is configured to handle this automatically.
The cmake-based build system assumes that you have Lapack and gfortran installed, see above instructions for more on that.
Build and install thermopack by running
mkdir build
cd build
cmake ..
make installThis will ensure that the thermopack dynamic library is properly installed to thermopack/installed and thermopack/addon/pycThermopack/thermopack.
To set up the python wrapper,
python addon/pycThermopack/map_platform_specifics.py
pip install addon/pycThermopack/this will generate the file addon/pycThermopack/thermopack/platform_specifics.py and install thermopack to your activated virtual environment.
ThermoPack can be configured to return computed properties as either tuples (v2) or using the Property struct (v3), this is toggled with
the -diffs flag when running map_platform_specifics.py as
python map_platform_specifics.py --diffs=v2 # Use tuples
python map_platform_specifics.py --diffs=v3 # use Propertythe default value is --diffs=v3. After running this command you should recieve a confirmation message that thermopack was successfully configured.
To compile thermopack (and Lapack) with Intel FORTRAN and MSVS, first run
git submodule update --init --recursive
from within the thermopack direcory, in order to clone Lapack. Then, run
mkdir build
cd build
cmake .. -DCMAKE_Fortran_COMPILER=ifort -DCMAKE_BUILD_TYPE=Release
cmake --build . --config=Release --target install
Compile and install Lapack, and install the thermopack dynamic library to thermopack/installed and thermopack/addon/pycThermopack/thermopack.
To configure and install the python-wrapper, run
python addon/pycThermopack/map_platform_specifics.py
pip install addon/pycThermopack/
Note: If your thermopack dynamic library is called libthermopack.dll, and not thermopack.dll, you will instead need to run
python addon/pycThermopack/map_platform_specifics.py --ifort=True
pip install addon/pycThermopack/
The Thermopack source code is downloaded by cloning the library to your local
computer. The following commands assume that you have a local installation of
Git, gfortran and
Python 3 with pip.
To compile using Intel FORTRAN, use make optim_ifort.
# Fetch and compile
git clone https://github.com/thermotools/thermopack.git
cd thermopack
make optim
# Prepare and install pycThermopack, aka "thermopack"
# Remark: On some systems, Python 3 is installed as python, not python3. If so,
# you can replace "python3" with "python" and perhaps also "pip3" with "pip" in
# the below.
cd addon/pycThermopack
python3 makescript.py optim
pip3 install --user .If you are working actively with the Thermopack code, either the Fortran
backend or the Python frontend, then it may be useful to install in editable
mode (aka develop mode). This will install a link to the develop files
instead of copying the files when installing. This can be done with the -e
option, i.e.:
pip3 install -e --user .See also addon/pycThermopack/README.md for more details on pycThermopack.
The easiest way to get started is via Homebrew. After
following the instructions on their website to set it up, install gcc
and make. Open a terminal (e.g. the default Terminal.app), and type:
brew install gcc make
Then follow the Linux instructions above (you may need to replace make with gmake).
Some additional packages like git and python can also be installed via
Homebrew before you start, but if you use a recent version of MacOS (e.g.
Catalina), then the versions installed by default should be sufficient.
Download and compile LAPACK and BLAS libraries (you will need CMake and a working compiler).
To be compatible with the current settings, you need to compile using Intel Fortran with Visual Studio, and configure as follows:
- Fortran/Data/Default Real KIND = 8
- Fortran/External Procedures/Calling Convention = cref
- Fortran/External Procedures/Name Case Interpretation = lowercase
- Fortran/External Procedures/String Length Argument Parsing = After All Arguments
- Fortran/External Procedures/Append Underscore to External Names = Yes
- Fortran/Floating Point/Floating Point Model = precise
Copy LAPACK and BLAS libraries to the paths:
- thermopack\lapack\lib\Debug
- thermopack\lapack\lib\Release
Open thermopack\MSVStudio\thermopack.sln using Visual Studio, and compile the wanted configuration.
See addon/pycThermopack/README.md for how to install pycThermopack.
Thermopack can also be compiled using gfortran in the MSYS2 environment. Download MSYS2 from https://www.msys2.org, and install and update the package system following the instructions given. Avoid having spaces in the MSYS2 installation directory path, as the Makefile might not work. Having a working MSYS2 installation, thermopack can be compiled after installing the following packages:
pacman -S git
pacman -S mingw-w64-x86_64-gcc-fortran
pacman -S mingw-w64-x86_64-openblas
pacman -S mingw-w64-x86_64-make
pacman -S mingw-w64-x86_64-dlfcnOpen the MSYS2 MinGW 64-bit application, and enter the following in the terminal:
git clone https://github.com/thermotools/thermopack.git
cd thermopack
mingw32-make.exe optimSee addon/pycThermopack/README.md for how to install pycThermopack for the MSYS2 environment.
See addon/docker/README.md for available Dockerfiles to run Thermopack with docker.
This is a short introduction to thermopack. Once you've gotten started, we recommend a look at the Examples in the GitHub repo. Comprehensive documentation for the methods available through the python interface can also be found in the doc page for the thermo class.. For more advanced users, a look at the more advanced page may also be useful.
Note: This guide applies to the most recent version of ThermoPack og GitHub. For guides applicable to versions found on PyPI, find the appropriate version in the sidebar on the ThermoPack homepage.
Equations of State (EoS's) in ThermoPack are classes. To do calculations for a given mixture an EoS object must first be initialized for that mixture, as demonstrated in the Initializing an EoS section. Then, a wide variety of thermodynamic computations can be done, as demonstrated in the remaining sections.
An overview of available equations of state can be found here.
An EoS is initialized by passing in the fluid identifiers of the mixture, for example
from thermopack.saftvrmie import saftvrmie
eos = saftvrmie('C1,CO2')will initialize a SAFT-VR Mie EoS for a mixture of methane and CO2. The complete list of component identifiers is in the Fluid identifiers list. PC-SAFT, SAFT-VRQ Mie and Lee-Kesler EoS are initialized in the same way, as
from thermopack import saftvrmie, saftvrqmie, pcsaft, lee_kesler
svrm = saftvrmie.saftvrmie('AR,KR') # SAFT-VR Mie EoS for Ar/Kr mixture
svrqm = saftvrqmie.saftvrqmie('HE') # SAFT-VRQ Mie EoS for pure He
pcs = pcsaft.pcsaft('BENZENE,NC6,NC12') # PC-SAFT EoS for ternary benzene/hexane/dodecane mixture
lk = lee_kesler.lee_kesler('N2,O2') # Lee-Kesler EoS for nitrogen/oxygen mixture For PC-SAFT, both the simplified PC-SAFT (SPC-SAFT) and Polar PC-SAFT (PCP-SAFT) are available
from thermopack.pcsaft import SPC_SAFT, PCP_SAFT
spcs = SPC_SAFT('NC6,NC12') # Simplified PC-SAFT
pcps = PCP_SAFT('H2O,MEOH') # Polar PC-SAFTThe cubic equations of state are found in the cubic module. Available cubic EoS's and more information on the individual cubics, mixing rules, etc. can be found on the cubic page.
from thermopack.cubic import SoaveRedlichKwong, RedlichKwong, PengRobinson, PengRobinson78
from thermopack.cubic import SchmidtWensel, PatelTeja, VanDerWaals
srk = SoaveRedlichKwong('NH3,C2') # SRK EoS for ammonia/ethane mixture
rk = RedlichKwong('NC6,CO2,NC12') # Redlich-Kwong EoS
pr = PengRobinson('IC4,NC10') # PR EoS for isobutane/decane mixture
pr78 = PengRobinson78('N2,O2,CO2') # # PR-78 EoS for isobutane/decane mixture
vdw = VanDerWaals('C1,C2,C3,N2,O2') # VdW EoS for methane/ethane/propane/nitrogen/oxygen mixture
sw = SchmidtWensel('R11,R12') # Schmidt-Wensel EoS for FCl3C/F2Cl2C mixture
pt = PatelTeja('PRLN') # Patel-Teja EoS for pure propyleneIn addition to these, the Translated-Consisten Peng-Robinson is available as
from thermopack.tcPR import tcPR
tcpr = tcPR('F6S,SO2') # Translated-Consistent PR EoS for SF6/SO2 mixtureFor more fine-tuned control of the cubic EoS, the parent class cubic can be initialised directly, to explicitly
set mixing rules, alpha-correlation etc.
Cubic-plus association EoS's are available for the SRK and PR EoS through the cpa module as
from thermopack.cpa import SRK_CPA, PR_CPA
srk_cpa = SRK_CPA('H2O,ETOH,PROP1OL') # SRK-CPA EoS for water/ethanol/propanol mixtureSeveral multiparameter EoS's can interfaced through the multiparameter.multiparam class. The available multiparameter EoS's are NIST-MEOS, MBWR16 and MBWR32. These are initialized as
from thermopack.multiparameter import multiparam
nist = multiparam('C3', 'NIST_MEOS') # NIST-MEOS EoS for propane
mbwr19 = multiparam('C1', 'MBWR19') # MBWR19 EoS for methane
mbwr32 = multiparam('C2', 'MBWR32') # MBWR32 EoS for ethaneplease note that not all fluids are supported for multiparameter equations of state, depending on what parameters are available in the fluid database.
Finally, the Extended-corresponding state EoS is available through the extended_csp.ext_csp class as
from thermopack.extended_csp import ext_csp
eos = ext_csp('C1,C2,C3,NC4', sh_eos='SRK', sh_alpha='Classic',
sh_mixing='vdW', ref_eos='NIST_MEOS', ref_comp='C3')For more information on the extended-csp EoS please see the Examples and the memo.
Now that we have an EoS initialized we can start computing stuff. The primary source on how to use individual methods in thermopack are the specific documentation of the thermo class. Here, a small subset of the functionality is demonstrated.
Note that all input is in SI units (moles/kelvin/pascal/cubic meters/joule)
Specific volume, given temperature, pressure and composition is computed as
from thermopack.saftvrmie import saftvrmie
eos = saftvrmie('NC6,NC10') # Hexane/decane mixture
T = 300 # Kelvin
p = 1e5 # Pascal
x = [0.2, 0.8] # Molar composition
vg, = eos.specific_volume(T, p, x, eos.VAPPH) # Molar volume of gas phase (NB: Notice the comma)
vl, = eos.specific_volume(T, p, x, eos.LIQPH) # Molar volume of liquid phase (NB: Notice the comma)where eos.VAPPH and eos.LIQPH are phase flags used to identify different phases. The commas are necessary because all output from thermopack methods are as tuples.
Similarly, pressure, internal energy, enthalpy, entropy, etc. and associated differentials can be computed via the methods chemical_potential_tv(T, V, n), internal_energy_tv(T, V, n), enthalpy_tv(T, V, n), helmholtz_tv(T, V, n), entropy_tv(T, V, n). For a full overview of the available property calculations see the TV-property interfaces and the Tp-property interfaces of the thermo class
If we want volume differentials, we use the same method, but set the flags to calculate differentials to True:
# Continued
vg, dvdp = eos.specific_volume(T, p, x, eos.VAPPH, dvdp=True) # Vapour phase molar volume and pressure differential
vl, dvdT = eos.specific_volume(T, p, x, eos.LIQPH, dvdt=True) # Liquid phase molar volume and temperature differential
_, dvdn = eos.specific_volume(T, p, x, eos.LIQPH, dvdn=True) # Liquid phase partial molar volumesDifferentials can be computed as functions of
# Continued
n_tot = 15 # Total number of moles
n = n_tot * x
H, dHdn_TV = eos.enthalpy_tv(T, vg, n, dhdn=True) # Compute enthalpy and derivative of enthalpy wrt. mole numbers at constant (T, V)
h_vap, dhvap_dn_Tp = eos.enthalpy(T, p, x, eos.VAPPH, dhdn=True) # Compute molar vapour phase enthalpy and derivative of molar vapour phase enthalpy wrt. mole numbers at constant (T, p)
h_liq, dliq_dn_Tp = eos.enthalpy(T, p, x, eos.LIQPH, dhdn=True) # Compute molar liquid phase enthalpy and derivative of molar liquid phase enthalpy wrt. mole numbers at constant (T, p)
H, dHdn_Tp = eos.enthalpy_tvp(T, vg, n, dhdn=True) # Compute enthalpy and derivative of enthalpy wrt. mole numbers at constant (T, p)Please note that heat capacities are not available directly, but must be computed as derivatives of enthalpy and internal energy, as
from thermopack.cubic import cubic
eos = cubic('C1,C3,NC6', 'SRK') # SRK EoS for a mixture of methane, propane and n-hexane
T = 300 # Kelvin
p = 1e5 # Pascal
x = [0.2, 0.1, 0.7] # Molar composition
_, Cp_vap = eos.enthalpy(T, p, x, eos.VAPPH, dhdt=True) # Vapour phase heat capacity at constant pressure, computed as (dH/dT)_{p,n}
_, Cp_liq = eos.enthalpy(T, p, x, eos.LIQPH, dhdt=True) # Liquid phase heat capacity at constant pressure, computed as (dH/dT)_{p,n}
vg, = eos.specific_volume(T, p, x, eos.VAPPH) # Computing vapour phase specific volume
vl, = eos.specific_volume(T, p, x, eos.LIQPH) # Liquid phase specific volume
_, Cv_vap = eos.internal_energy_tv(T, vg, x, dedt=True) # Vapour phase heat capacity at constant volume, computed as (dU/dT)_{V,n}
_, Cv_liq = eos.internal_energy_tv(T, vl, x, dedt=True) # Liquid phase heat capacity at constant volume, computed as (dU/dT)_{V,n}As with other calculations, the primary source on how available methods for flash- and equilibria calculations and how to use them is the documentation of the thermo class.. Here we give a short introduction, for more extensive examples see the pyExamples directory.
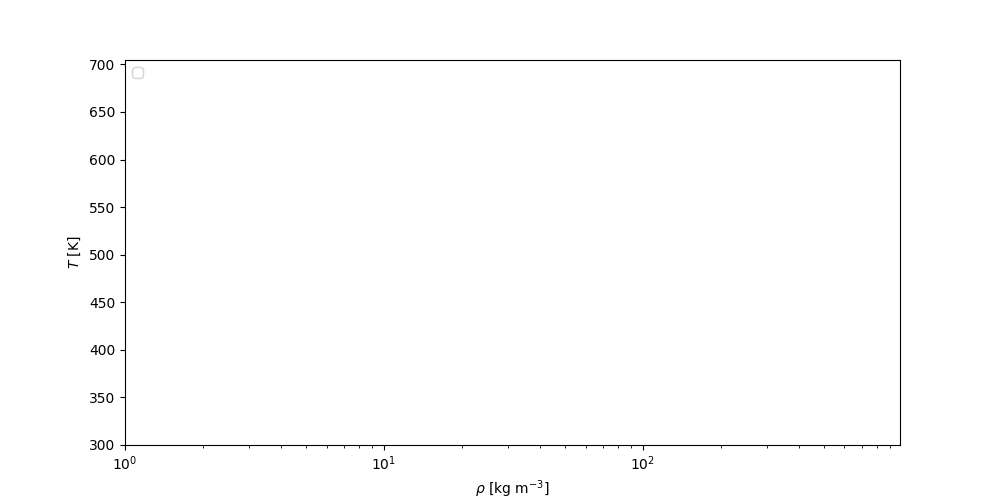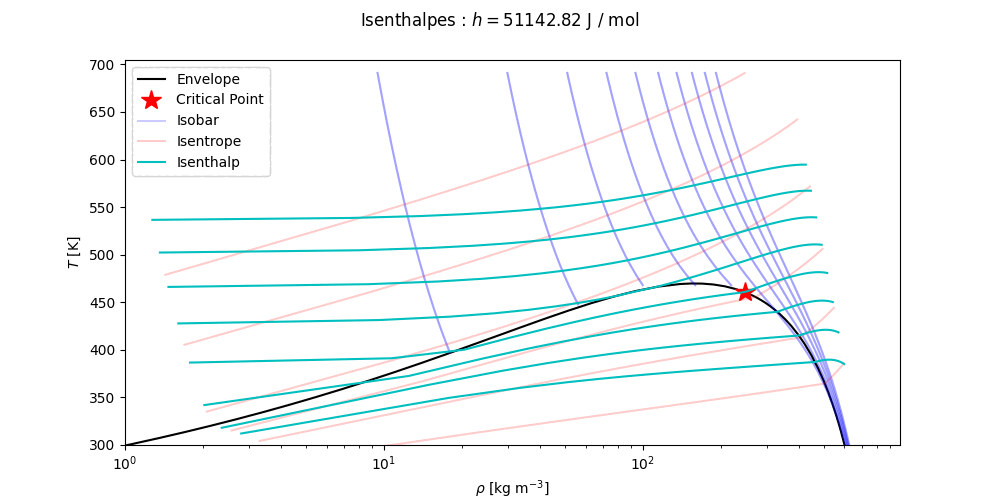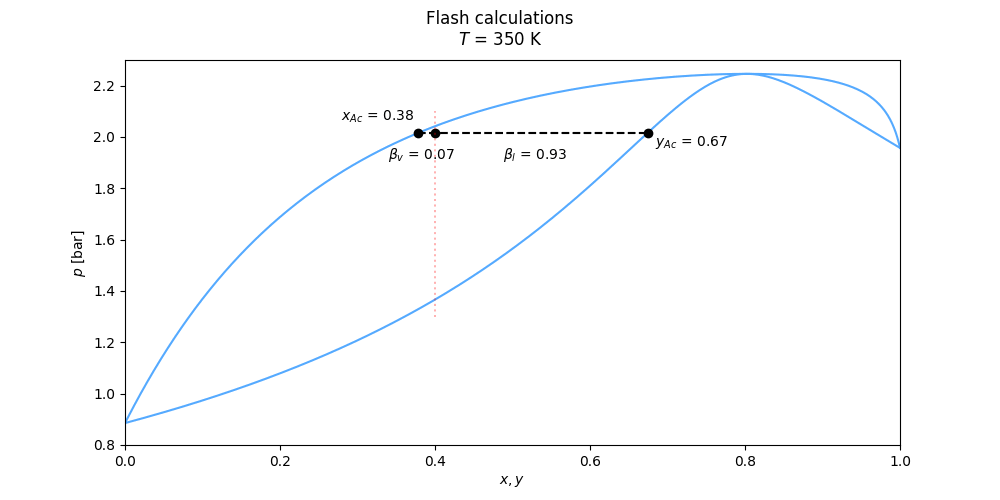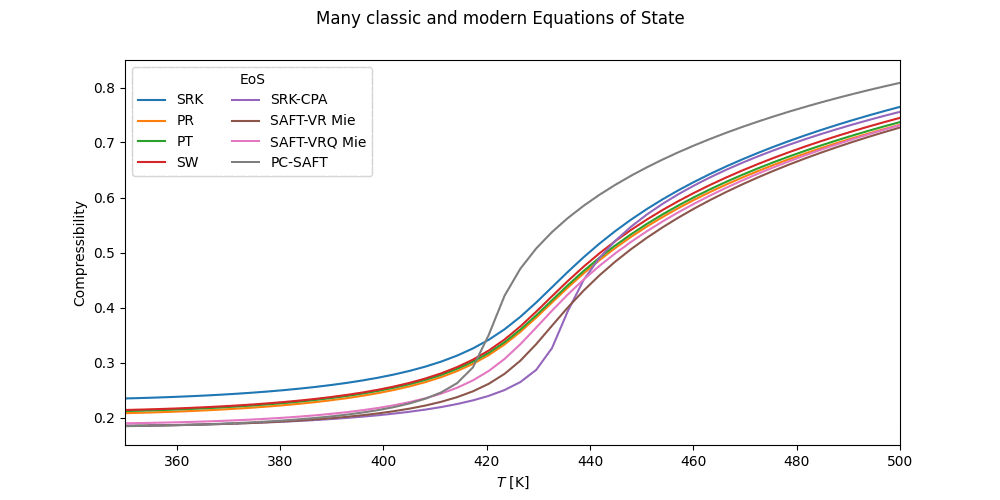
Flash calculations of several kinds are handled by the methods twophase_tpflash(), twophase_psflash(), twophase_phflash() and twophase_uvflash().
See the Flash interfaces in the documentation of the thermo class for the specifics on the different flash routines.
An example calculation using twophase_tpflash() may be done as
from thermopack.saftvrqmie import saftvrqmie
# SAFT-VRQ Mie for Hydrogen/Helium/Neon mixture
eos = saftvrqmie('H2,HE,NE', minimum_temperature=20) # NB: Set minimum temperature low enough when working at very low temperatures
T = 35 # Kelvin
p = 3e6 # Pascal (30 bar)
z = [0.1, 0.25, 0.65] # Molar composition
flsh = eos.two_phase_tpflash(T, p, z) # flsh is a FlashResult object
print(flsh)
### Output: ###
# FlashResult object for Tp-flash
# Containing the attributes (description, name, value):
# Flash type flash_type : Tp
# Total composition z : [0.1, 0.25, 0.65]
# Temperature [K] T : 35
# pressure [Pa] p : 3000000.0
# Liquid phase composition x : [0.05407302 0.03859287 0.90733411]
# Vapour phase composition y : [0.14642524 0.46370066 0.3898741 ]
# Vapour fraction betaV : 0.497302408174766
# Liquid fraction betaL : 0.5026975918252341
# Phase indentifier index phase : 0 the result of the flash is accessed from the attributes of the FlashResult object, found in utils.py, as
# Continued
x = flsh.x # Liquid composition
y = flsh.y # Vapour composition
betaL = flsh.betaL # Liquid fraction
# ... etcThe FlashResult object returned by the different flash routines all contain the same attributes.
ThermoPack has interfaces to trace (T,p)-, (T,v)- and (p,x,y)-phase envelopes. For the full documentation, see the docs of the thermo class. For more comprehensive examples, see the Examples.
Phase envelopes can be generated directly with the method get_envelope_twophase() as
# Continued
T, p = eos.get_envelope_twophase(1e5, x) # arrays of temperature and pressure for phase envelope, starting at 1 bar.
plt.plot(p, T) # Tp-projection of phase envelope
T, p, v = eos.get_envelope_twophase(1e5, x, calc_v=True) # Also return the specific volume at each point along the phase envelope
plt.plot(1 / v, T) # rho-T projection of the phase envelopeTo compute pxy-type phase envelopes, we use the get_binary_pxy() method. This method returns a XYDiagram struct, which
holds the composition and pressure / temperature of each equilibrium phase.
The XYDiagram struct has the attributes lle, l1ve and l2ve, where
lleholds the composition and pressure / temperature of the Liquid 1 - Liquid 2 equilibrialle.x1is the composition of Liquid 1 (mole fraction of species 1)lle.x2is the composition of Liquid 2 (mole fraction of species 1)lle.p/lle.Tis the pressure / temperature along the phase boundary
l1veholds the composition and pressure / temperature of the Liquid 1 - Vapour equilibrial1ve.xis the composition of Liquid 1 (mole fraction of species 1)l1ve.yis the composition of the vapour (mole fraction of species 1)l1ve.p/l1ve.Tis the pressure / temperature along the phase boundary
l2veholds the composition and pressure / temperature of the Liquid 2 - Vapour equilibrial2ve.xis the composition of Liquid 2 (mole fraction of species 1)l2ve.yis the composition of the vapour (mole fraction of species 1)l2ve.p/l2ve.Tis the pressure / temperature along the phase boundary
We get the phase diagram as
from thermopack.cpa import SRK_CPA
eos = SRK_CPA('NC6,H2O') # CPA-SRK eos for Hexane/water mixture
T = 350
pxy = eos.get_binary_pxy(T) # Returns a XYDiagram struct
print(pxy)
# Result:
# XYDiagram object with attributes (name : description)
# lle : Liquid 1 - Liquid 2 Equilibrium
# PxyEquilibrium object with attributes (description, name, value)
# Type of equilibrium type : lle
# Liquid 1 mole fraction, species 1 x1 : [4.278e-07 ... 4.145e-07], len(x1) = 457
# Liquid 2 mole fraction, species 1 x2 : [9.969e-01 ... 9.974e-01], len(x2) = 457
# Pressure p : [1.648e+05 ... 1.500e+07], len(p) = 457
#
# l1ve : Liquid 1 - Vapour Equilibrium
# PxyEquilibrium object with attributes (description, name, value)
# Type of equilibrium type : lve
# Liquid mole fraction, species 1 x : [4.278e-07 ... 2.197e-07], len(x) = 66
# Vapour mole fraction, species 1 y : [7.880e-01 ... 6.461e-01], len(y) = 66
# Pressure p : [1.648e+05 ... 1.000e+05], len(p) = 66
#
# l2ve : Liquid 2 - Vapour Equilibrium
# PxyEquilibrium object with attributes (description, name, value)
# Type of equilibrium type : lve
# Liquid mole fraction, species 1 x : [9.969e-01 ... 1.000e+00], len(x) = 100
# Vapour mole fraction, species 1 y : [7.880e-01 ... 1.000e+00], len(y) = 100
# Pressure p : [1.648e+05 ... 1.289e+05], len(p) = 100For ease of use, the XYDiagram struct is iterable, such that we can unpack it efficiently as
lle, l1ve, l2ve = pxy
Now, we can plot the phase diagram as
# Continued
import matplotlib.pyplot as plt
# Liquid-liquid phase boundaries
# pxy.lle holds the composition and pressure of the liquid phases
plt.plot(pxy.lle.x1, pxy.lle.p, label='Liquid 1 composition') # lle.x1 is the mole fraction of component 1 (NC12) in Liquid 1 along the phase boundary
plt.plot(pxy.lle.x2, pxy.lle.p, label='Liquid 2 composition') # lle.x2 is the mole fraction of component 1 (NC12) in Liquid 2 along the phase boundary
# Liquid 1-vapour phase boundaries
# pxy.l2ve holds composition and pressure along the Liquid 1 - Vapour phase boundary
plt.plot(pxy.l1ve.x, pxy.l1ve.p, label='Liquid 1 bubble line') # l1ve.x is the mole fraction of component 1 (NC12) in Liquid 1 along the Liquid 1 - Vapour phase boundary
plt.plot(pxy.l1ve.y, pxy.l1ve.p, label='Liquid 1 dew line') # l1ve.y is the mole fraction of component 1 (NC12) in the Vapour phase along the Liquid 1 - Vapour phase boundary
# Liquid 2-vapour phase boundaries
# L2VE[2] is the pressure along the Liquid 2 - Vapour phase boundary
plt.plot(pxy.l2ve.x, pxy.l2ve.p, label='Liquid 2 bubble line') # l2ve.x is the mole fraction of component 1 (NC12) in Liquid 2 along the Liquid 2 - Vapour phase boundary
plt.plot(pxy.l2ve.y, pxy.l2ve.p, label='Liquid 2 dew line') # l2ve.y is the mole fraction of component 1 (NC12) in the Vapour phase along the Liquid 2 - Vapour phase boundary
plt.ylabel('Pressure [Pa]') # The third element in each tuple is the pressure along the phase boundary
plt.xlabel('Molar composition')The method get_binary_txy works in the same way, only replacing the p attribute with T, such that we can compute
p = 1e5
lle, l1ve, l2ve = eos.get_binary_txy(p) # Unpacking the XYDiagram
# Liquid-liquid phase boundaries
# l1ve holds the composition and pressure of Liquid 1 and Vapour along the phase boundary
plt.plot(l1ve.x, l1ve.T, label='Liquid 1 composition') # l1ve.x is the mole fraction of component 1 (NC12) in Liquid 1 along the phase boundary
plt.plot(l1ve.y, l1ve.T, label='Vapour composition') # l1ve.y is the mole fraction of component 1 (NC12) in Vapour along the phase boundary
# ... etc ...
If an equilibrium is not found, for example there is only one vapour-liquid equilibria, and no liquid-liquid equilibria, the arrays corresponding to the non-existent equilibria are empty, i.e.
T = 300
pxy = get_binary_pxy(T) # Some mixture that has only one vapour - liquid equilibria at 300 K
print(len(pxy.lle.x1) == 0) # True
print(len(pxy.lle.x2) == 0) # True
print(len(pxy.lle.p) == 0) # True
print(len(pxy.l1ve.x) == 0) # False
print(len(pxy.l1ve.y) == 0) # False
print(len(pxy.l1ve.p) == 0) # False
print(len(pxy.l2ve.x) == 0) # True
print(len(pxy.l2ve.y) == 0) # True
print(len(pxy.l2ve.p) == 0) # True
We can also compute the bubble-temperature, pressure etc. directly using the methods bubble_temperature(p, z), bubble_pressure(T, z), dew_temperature(p, z) and dew_pressure(T, z), where z is the composition of the mixture, as
eos = cubic('CO2,C1', 'SRK')
x = [0.5, 0.5] # Total composition of the mixture
p_dew, y_dew = eos.dew_pressure(250, x) # Calculates dew pressure and dew composition at 250 K
T_dew, y_dew = eos.dew_temperature(1e5, x) # Calculates dew temperature and dew composition at 1 bar
p_bub, x_bub = eos.bubble_pressure(230, x) # Calculates bubble pressure and bubble composition at 230 K
T_bub, x_bub = eos.bubble_temperature(1e5, x) # Calculates bubble temperature and bubble composition at 1 barVarious isolines can be computed using the methods get_isotherm, get_isobar, get_isentrope and get_isenthalp. In the following code snippet, the default values of the keyword arguments are indicated.
from thermopack.pcsaft import pcsaft
eos = pcsaft('NC6,NC12')
x = [0.2, 0.8]
# Calculate pressure, specific volume, specific entropy and specific enthalpy along the isotherm at 300 K
# from p = minimum_pressure to p = maximum_pressure. Compute at most nmax points.
p_iso_T, v_iso_T, s_iso_T, h_iso_T = eos.get_isotherm(300, x, minimum_pressure=1e5, maximum_pressure=1.5e7, nmax=100)
# Calculate temperature, specific volume, specific entropy and specific enthalpy along the isobar at 1 bar
# from T = minimum_temperature to T = maximum_temperature. Compute at most nmax points.
T_iso_p, v_iso_p, s_iso_p, h_iso_p = eos.get_isobar(1e5, x, minimum_temperature=200, maximum_temperature=500, nmax=100)
# Calculate temperature, pressure, specific volume and specific entropy along the isenthalp at 1 kJ / mol
# Start at the upper of (minimum_pressure, minimum_temperature)
# End at the lower of (maximum_pressure, maximum_temperature)
T_iso_h, p_iso_h, v_iso_h, s_iso_h = eos.get_isenthalp(1e3, x, minimum_pressure=1e5, maximum_pressure=1.5e7,
minimum_temperature=200, maximum_temperature=500,
nmax=100)
# Calculate temperature, pressure, specific volume and specific enthalpy along the isentrope at 5 J / mol K
# Start at the upper of (minimum_pressure, minimum_temperature)
# End at the lower of (maximum_pressure, maximum_temperature)
T_iso_s, p_iso_s, v_iso_s, h_iso_s = eos.get_isentrope(5, x, minimum_pressure=1e5, maximum_pressure=1.5e7,
minimum_temperature=200, maximum_temperature=500,
nmax=100)Thermopack has a critical point solver, which is called as
eos = saftvrqmie('HE,NE') # Use FH-corrected Mie potentials for Helium calculations!
n = [5, 10]
Tc, Vc, pc = eos.critical(n) # Compute the critical temperature, pressure and volume given mole numbers
vc = Vc / sum(n) # Critical specific volume computed from critical volume and mole numbers.The solver accepts initial guesses for the critical values through the kwargs temp, and v. The error tolerance can be set via the tol kwarg (default is tol=1e-7).
In thermopack we're able to both set and get a wide array of coefficients and parameters depending on the models we are utilizing.
Setting and getting the attractive energy interaction parameter $k_{ij}$ and co-volume interaction parameter $l_{ij}$
Starting with the attractive energy interaction parameter (kij). The parameter can be set using the function set_kij after initialising the equation and state. The function requires that you first write in the number of the components and subsequently the new interaction parameter i.e. (component number 1, component number 2, new kij value). If we're curious as to what parameter the EOS is already using we can see this by using the function get_kij which returns the value as a float given the component numbers as input i.e. (component number 1, component number 2).
cs = cubic('CO2,N2',"SRK","Classic","Classic")
#We set the interaction parameter to be -0.032
cs.set_kij(1,2,-0.032)
#We want to see what the interaction parameter is which returns that kij = -0.032
kij = cs.get_kij(1,2)The procedure for setting and getting co-volume interaction parameters is analogous to the getting and setting of attractive energy parameters. Simply use the functions set_lij and get_lij instead.
#We set the parameter to be -0.032
cs.set_lij(1,2,-0.032)
#We want to see what the parameter is which returns that lij = -0.032
lij = cs.get_lij(1,2)Cubic Equations of state implemented in ThermoPack can be accessed through the generic cubic class.
This class also offers a variety of methods to tune the alpha-function, mixing rules etc. See the documentation for
the cubic class for more information.
Property calculations in ThermoPack can be done either through the TV-interfaces, the Tp-interfaces or the TVp-interfaces.
The difference between the TV- and Tp- interface is only what variables the properties are computed as functions of, and what variables are held constant in the derivatives. TV-interface methods compute properties as functions of
The TVp-interface methods on the other hand take
import numpy as np
from thermopack.cubic import cubic
eos = cubic('O2,N2', 'PR') # PR EoS for O2/N2 mixture
T = 300 # Kelvin
p = 1e5 # Pascal
x = np.array([0.21, 0.79]) # Molar composition (of air)
n_tot = 10 # Total number of moles
n = n_tot * x # Vector of mole numbers
v, = eos.specific_volume(T, p, x, eos.VAPPH) # Compute specific volume of vapour phase
V = v * n_tot # Total volume
# Computing the TOTAL Enthalpy (J), given (T, V, n)
# The differentials are computed for H = H(T, V, n), with
# "subscripts" indicating the variables held constant
H_tvn, dHdT_Vn, dHdn_TV = eos.enthalpy_tv(T, V, n, dhdt=True, dhdn=True)
# Computing the SPECIFIC VAPOUR phase enthalpy (J / mol), given (T, p, n)
# The differentials are computed for h_vap = h_vap(T, p, n), with the
# "subscripts" indicating the variables held constant
h_vap_tpn, dh_vap_dt_pn, dh_vap_dn_Tp = eos.enthalpy(T, p, n, eos.VAPPH, dhdt=True, dhdn=True)
# Computing the SPECIFIC LIQUID phase enthalpy (J / mol), given (T, p, n)
# The differentials are computed for h_liq = h_liq(T, p, n), with the
# "subscripts" indicating the variables held constant
h_liq_tpn, dh_liq_dt_pn, dh_liq_dn_Tp = eos.enthalpy(T, p, n, eos.LIQPH, dhdt=True, dhdn=True)
# Computing the TOTAL Enthalpy (J), given (T, V, n)
# NOTE : The differentials are computed for H = H(T, p, n)
# NOT for H = H(T, V, n)
H_tpn, dHdt_pn, dHdn_Tp = eos.enthalpy_tvp(T, V, n, dhdt=True, dhdn=True)Besides enthalpy_tvp, there are currently available TVp-interfaces for entropy_tvp and thermo_tvp (logarithm of fugacity coefficients).
The fluid database consists of a set of
.json-files in the
fluids directory. These files are
are used to auto-generate the FORTRAN-files
compdatadb.f90 and
saftvrmie_datadb.f90 by running
the respective python scripts
compdata.py and
saftvrmie.py found in the
directory addon/pyUtils/datadb/.
The files are generated in the current working directory and must be
copied to the src-directory
before recompiling ThermoPack to make the fluids available.
A <fluid\>.json file must
contain a minimal set of data to be valid. This includes the critical
point, accentric factor, mole weight and ideal gas heat capacity.
Several different correlations for the heat capacity are available, selected by the "correlation"-key in the "ideal-heat-capacity-1" field of the fluid files. These are summarized in the table below.
Ideal gas heat capacity correlations, and the corresponding keys used in the fluid-database.
| Key | Correlation | Equation | Unit |
|---|---|---|---|
| 1 | Sherwood, Reid & Prausnitz(a) | ||
| 2 | API-Project | 44 | - |
| 3 | Hypothetic components | - | - |
| 4 | Sherwood, Reid & Prausnitz(b) | ||
| 5 | Ici (Krister Strøm) | ||
| 6 | Chen, Bender (Petter Nekså) | ||
| 7 | Aiche, Daubert & Danner(c) | ||
| 8 | Poling, Prausnitz & O’Connel(d) | ||
| 9 | Linear function and fraction | ||
| 10 | Leachman & Valenta for H2 | - | - |
| 11 | Use TREND model | - | - |
| 12 | Shomate equation |
||
| 13 | Einstein equation sum |
(a)3rd ed.(c)DIPPR-database
(b)4th ed.(d)5th ed.
<fluid\>.json file, while Methane.json file for an working example of both the melting_curve and sublimation_curve parameters.
The reduced temperature used in the correlations is defined as
The last character in the correlation string defines how the reducing pressure combines with
For the melting curve calculation
For the sublimation curve calculation
The melting/sublimation curves can be scaled to match the saturation pressure at the triple temperature,
In order to specify fluids in Thermopack you need to use fluid identifiers as shown in the table below. The 'SAFT-VR', 'PC-SAFT' and 'CPA' columns indicate which fluids SAFT-EoS and CPA parameters are available for.
You may have to scroll right to view the whole table.
| Fluid name | CAS Number | Fluid identifyer | SAFT-VR | PC-SAFT | CPA |
|---|---|---|---|---|---|
| 1,1,1,2-Tetrafluoroethane | 811-97-2 | R134a | |||
| 1,1,1-Trifluoroethane | 420-46-2 | R143a | |||
| 1,1-Difluoroethane | 75-37-6 | R152a | |||
| 1,1-Difluoroethylene | 75-38-7 | R1132a | |||
| 1,2-Dichlorotetrafluoroethane | 76-14-2 | R114 | |||
| 1,3-Butadiene | 106-99-0 | 13BD | |||
| 1-Butanol | 71-36-3 | BUT1OL | ✔ | ✔ | |
| 1-Chloro-1,1,2,2-tetrafluoroethane | 354-25-6 | R124a | |||
| 1-Chloro-1,1-difluoroethane | 75-68-3 | R142b | |||
| 1-Hexanol | 111-27-3 | HEX1OL | ✔ | ✔ | |
| 1-Pentanol | 71-41-0 | PENT1OL | ✔ | ✔ | |
| 1-Propanol | 71-23-8 | PROP1OL | ✔ | ✔ | |
| 2,3,3,3-Tetrafluoropropene | 754-12-1 | R1234yf | |||
| 2-Chloro-1,1,1,2-tetrafluoroethane | 2837-89-0 | R124 | |||
| 2-Methylhexane | 591-76-4 | 2MHX | |||
| 3-Methylpentane | 96-14-0 | 3MP | |||
| Acetone | 67-64-1 | ACETONE | ✔ | ||
| Acetylene | 74-86-2 | ACETYLENE | ✔ | ||
| Ammonia | 7664-41-7 | NH3 | ✔ | ✔ | ✔ |
| Argon | 7440-37-1 | AR | ✔ | ✔ | |
| Benzene | 71-43-2 | BENZENE | ✔ | ||
| Butanal | 123-72-8 | BUTANAL | ✔ | ||
| Carbon dioxide | 124-38-9 | CO2 | ✔ | ✔ | ✔ |
| Carbon monoxide | 630-08-0 | CO | |||
| Carbon tetrafluoride | 75-73-0 | R14 | |||
| Chlorine | 7782-50-5 | CL2 | ✔ | ||
| Chlorodifluoromethane | 75-45-6 | R22 | |||
| Chloropentafluoroethane | 76-15-3 | R115 | |||
| Chlorotrifluoromethane | 75-72-9 | R13 | |||
| Chlorotrifluorosilane | 14049-36-6 | ClF3Si | |||
| Cyclohexane | 110-82-7 | CYCLOHEX | ✔ | ||
| Cyclopropane | 75-19-4 | C3_1 | |||
| Deuterium | 7782-39-0 | D2 | ✔ | ||
| Di-methyl ether | 115-10-6 | DME | ✔ | ||
| Di-n-hexyl ether | 112-58-3 | S434 | |||
| Dichlorodifluoromethane | 75-71-8 | R12 | |||
| Dichlorofluoromethane | 75-43-4 | R21 | |||
| Difluoromethane | 75-10-5 | R32 | |||
| Dinitrogen tetroxide | 10544-72-6 | N2O4 | |||
| Equilibrium-hydrogen | 1333-74-0 | E-H2 | |||
| Ethane | 74-84-0 | C2 | ✔ | ✔ | |
| Ethanol | 64-17-5 | ETOH | ✔ | ✔ | ✔ |
| Ethylbenzene | 100-41-4 | EBZN | |||
| Ethylene glycol | 107-21-1 | MEG | |||
| Ethylene | 74-85-1 | C2_1 | ✔ | ||
| Helium-4 | 7440-59-7 | HE | ✔ | ||
| Hexafluoroethane | 76-16-4 | R116 | |||
| Hydrazine | 302-01-2 | N2H4 | ✔ | ||
| Hydrogen peroxide | 7722-84-1 | H2O2 | |||
| Hydrogen sulfide | 7783-06-4 | H2S | ✔ | ✔ | |
| Hydrogen | 1333-74-0 | H2 | ✔ | ||
| Isobutane | 75-28-5 | IC4 | ✔ | ||
| Isopentane | 78-78-4 | IC5 | ✔ | ||
| Krypton | 7439-90-9 | KR | ✔ | ✔ | |
| Lennard-jones_fluid | LJF | ✔ | |||
| Methane | 74-82-8 | C1 | ✔ | ✔ | |
| Methanol | 67-56-1 | MEOH | ✔ | ✔ | ✔ |
| Methyl fluoride | 593-53-3 | R41 | |||
| Methylcyclopentane | 96-37-7 | MTC5 | |||
| Neon | 7440-01-9 | NE | ✔ | ||
| Nitric oxide | 10102-43-9 | NO | |||
| Nitrogen | 7727-37-9 | N2 | ✔ | ✔ | |
| Nitrous oxide | 10024-97-2 | N2O | |||
| Octafluoropropane | 76-19-7 | R218 | |||
| Ortho-hydrogen | 1333-74-0 | O-H2 | ✔ | ||
| Oxygen | 7782-44-7 | O2 | ✔ | ✔ | |
| Para-hydrogen | 1333-74-0 | P-H2 | ✔ | ||
| Pentafluoroethane | 354-33-6 | R125 | |||
| Propadiene | 463-49-0 | ALLENE | |||
| Propane | 74-98-6 | C3 | ✔ | ✔ | ✔ |
| Propylene | 115-07-1 | PRLN | |||
| Pseudo | XXX | PSEUDO | |||
| Sulfur dioxide | 7446-09-5 | SO2 | |||
| Sulfur hexafluoride | 2551-62-4 | F6S | |||
| Tetrafluoroethylene | 116-14-3 | R1114 | |||
| Tetrafluorohydrazine | 10036-47-2 | F4N2 | |||
| Toluene | 108-88-3 | TOLU | ✔ | ||
| Trans-1,3,3,3-tetrafluoropropene | 29118-24-9 | R1234ze | |||
| Trichlorofluoromethane | 75-69-4 | R11 | |||
| Trifluoroamine oxide | 13847-65-9 | F3NO | |||
| Trifluoromethane | 75-46-7 | R23 | |||
| Water | 7732-18 | H2O | ✔ | ✔ | ✔ |
| Xenon | 7440-63-3 | XE | ✔ | ||
| m-Xylene | 108-38-3 | MXYL | |||
| n-Butane | 106-97-8 | NC4 | ✔ | ✔ | ✔ |
| n-Decane | 124-18-5 | NC10 | ✔ | ✔ | ✔ |
| n-Docosane | 629-97-0 | NC22 | ✔ | ✔ | |
| n-Dodecane | 112-40-3 | NC12 | ✔ | ||
| n-Eicosane | 112-95-8 | NC20 | ✔ | ✔ | |
| n-Heneicosane | 629-94-7 | NC21 | ✔ | ||
| n-Heptadecane | 629-78-7 | NC17 | ✔ | ||
| n-Heptane | 142-82-5 | NC7 | ✔ | ✔ | ✔ |
| n-Hexadecane | 544-76-3 | NC16 | ✔ | ||
| n-Hexane | 110-54-3 | NC6 | ✔ | ✔ | ✔ |
| n-Hydrogen | 1333-74-0 | N-H2 | |||
| n-Nonadecane | 629-92-5 | NC19 | ✔ | ||
| n-Nonane | 111-84-2 | NC9 | ✔ | ✔ | ✔ |
| n-Octadecane | 593-45-3 | NC18 | ✔ | ||
| n-Octane | 111-65-9 | NC8 | ✔ | ✔ | ✔ |
| n-Pentacosane | 629-99-2 | NC25 | ✔ | ||
| n-Pentadecane | 629-62-9 | NC15 | ✔ | ✔ | |
| n-Pentan | 109-66-0 | NC5 | ✔ | ✔ | ✔ |
| n-Tetracosane | 646-31-1 | NC24 | ✔ | ||
| n-Tetradecane | 629-59-4 | NC14 | ✔ | ||
| n-Tricosane | 638-67-5 | NC23 | ✔ | ||
| n-Tridecane | 629-50-5 | NC13 | ✔ | ||
| n-Undecane | 1120-21-4 | NC11 | ✔ | ||
| o-Xylene | 95-47-6 | OXYL | |||
| p-Xylene | 106-42-3 | PXYL |